US Pharm. 2014;39(2):HS2-HS10.
ABSTRACT: Great advances have been made in the arena of cancer drug therapy. A number of modalities, such as targeted therapies, have emerged that have drastically changed the clinical landscape of cancer-related drug therapy. The advent of these various targeted therapies, however, has increased the incidence of cardiotoxicity-related events in cancer patients. It is crucial that cardiotoxicity in cancer patients be managed effectively by the healthcare practitioner in order to decrease the mortality and morbidity associated with the underlying malignancy.
The goals of cancer drug therapy are to cure the disease, prevent disease recurrence, and decrease mortality. More cancer patients are surviving because of advances in cancer-related drug therapies. Cardiotoxicity, in which the heart muscle is damaged and cannot efficiently pump blood to oxygenate the body’s vital organs, is one of the most prominent morbidities in cancer patients surviving 5 to 10 years post therapy. Cardiotoxicity may lead to increased mortality and disease progression because the therapeutic options are suboptimal.1,2
There are many deficits in scientific knowledge concerning cancer therapy–induced cardiotoxicity, including the area of basic clinical science. Such deficits encumber progress in this area. Major cancer-related therapies associated with cardiac dysfunction include the anthracyclines and trastuzumab (Herceptin), the monoclonal antibody (Mab) targeted against human epidermal growth factor receptor (HER2)/neu.1,2 This article will examine the most common cardiotoxic cancer-related drug therapies.
ANTHRACYCLINES
The anthracyclines, which include doxorubicin, daunorubicin, epirubicin, idarubicin, and valrubicin, are chemotherapeutic antibiotics that inhibit enzyme topoisomerase II. This inhibition prevents DNA and RNA from fusing by the intercalation of DNA base pairs and steric hindrance. Doxorubicin (Adriamycin), one of the major anthracyclines used in chemotherapy (ChT), is indicated to treat many different cancers, including breast cancer (BC), acute lymphocytic leukemia (ALL), lymphoma, acute myeloid leukemia, soft-tissue and bone sarcoma, ovarian cancer, gastric cancer, bladder cancer, neuroblastoma, Wilms’ tumor, small cell lung cancer (SCLC), and thyroid cancer.3-9
Anthracycline-Associated Cardiotoxicity
Anthracyclines are known for their potent cardiotoxic effects, predominantly heart failure (HF) and cardiomyopathies. Baseline and periodic measurements of left ventricular ejection fraction (LVEF) are therefore essential. A multiple gated acquisition (MUGA) scan or echocardiogram (ECHO) should be performed at baseline for proper assessment of LVEF. MUGA, a noninvasive procedure used to measure cardiac function, can locate the part of the heart muscle that has been damaged and evaluate the type of damage. ECHO uses sound waves to produce images of the heart to determine how fast the heart is beating and pumping blood.3-9
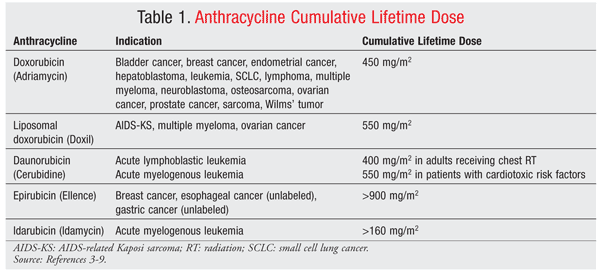
Anthracycline-associated cardiotoxicity (AAC) is cumulative and permanent. The three types of AAC are immediate, early-onset, and chronic progressive cardiotoxicity. The primary risk factor for AAC is the cumulative lifetime dose (CLD) of the anthracycline. The CLD must be documented and retained for proper drug dosage, patient safety, and positive therapeutic outcomes. Patients are predisposed to the development of congestive heart failure (CHF) when the CLD is exceeded (TABLE 1).3-9
The main mechanism of action (MOA) for AAC is hypothesized to be the generation of oxygen free radicals, which cause oxidative stress, resulting in cellular damage in the cardiomyocyte and eventually apoptosis, which is directly correlated with the dose of anthracycline administered. Iron accumulation occurs through both enzymatic and nonenzymatic channels precipitated by free-radical generation and redox-associated injury to the cardiomyocyte. As mitochondrial damage occurs, calcium levels rise, inhibiting the action of the sarcoplasmic reticulum and decreasing the function of the sodium-potassium pump. All of these factors are implicated in AAC.3-9
Risk factors for AAC include prior anthracycline therapy, the drug’s CLD, younger age at treatment initiation, current pregnancy, doxorubicin dose ≥300 mg/m2, epirubicin ≥600 mg/m2, mediastinal radiation (RT), combination ChT regimens, and increased overall survival.3-8
Carvedilol (Coreg), a nonselective beta-blocker providing adrenergic blockade, is approved to treat HF, hypertension (HTN), and impaired left ventricular function following myocardial infarction (MI). Clinical studies have evaluated its use in the prevention of anthracycline-induced cardiomyopathy. Subjects received either placebo or carvedilol 12.5 mg once daily prior to the start of ChT, and the primary endpoint was systolic function. There was less mortality in the carvedilol group, but the results were nonsignificant.10
Administration of anthracyclines to adult patients via slow infusion versus bolus causes lower drug peak levels and reduces the pharmacokinetic factor of cardiotoxicity. Decreasing the peak drug level of an anthracycline has a cardioprotective effect. Other measures for reducing AAC include liposomal anthracycline preparations and dexrazoxane (Zinecard).11
Liposomal Anthracyclines
In these agents, liposomes—which have a bilayer phospholipid covering—are used to encapsulate the anthracycline, mitigating the potential for cardiac-related drug-induced damage. The liposomal encapsulation alters the pharmacokinetics and tissue distribution of doxorubicin, and the liposome increases the half-life of the anthracycline preparation. The bilayer phospholipid covering reduces the amount of drug exposed to heart muscle, and the slow release of anthracycline prevents high peak plasma levels of the drug.12-20
Liposomal agents include liposomal daunorubicin (DaunoXome), pegylated liposomal doxorubicin (Caelyx, Doxil), and liposomal doxorubicin (Myocet). These products do not possess the identical amount of lipid, and the addition of polyethylene glycol to the liposome alters the pharmacodynamics of the agent. The pegylated product protects the anthracycline from phagocytosis. Liposomal agents should not be substituted for the conventional form of the anthracycline.12-20
Liposomal daunorubicin has exhibited cardiotoxicity at cumulative doses of 600 to 900 mg/m2. Currently, insufficient published data are available to assess the cardiotoxicity of liposomal daunorubicin versus conventional daunorubicin.12-20
Liposomal doxorubicin is associated with a lower rate of cardiotoxic events compared with conventional doxorubicin. The clinical activity of liposomal doxorubicin is comparable to that of nonliposomal doxorubicin. The pegylated liposomal form of doxorubicin has the lowest rate of cardiotoxicity associated with therapy. At a cumulative dose of >500 mg/m2, pegylated liposomal doxorubicin had a much lower rate of cardiotoxicity than nonliposomal doxorubicin. Patients with LVEF <50% should not receive liposomal anthracycline therapy. When the cumulative dose of the liposomal anthracycline reaches 400 mg/m2, MUGA should be performed and repeated thereafter for cumulative dose increases of 100 to 120 mg/m2. If clinical cardiotoxicity develops, the liposomal preparation should be discontinued. If any type of cardiac decompensation is noted, a diagnostic heart evaluation should be performed.12-20
CARDIOPROTECTIVE AGENTS
Dexrazoxane is a cardioprotective agent used in women with metastatic BC who have received a cumulative dose of doxorubicin of ≥300 mg/m2. It is given at a 10:1 ratio (e.g., dexrazoxane 500 mg/m2:doxorubicin 50 mg/m2) to prevent cardiomyopathies. Dexrazoxane reduces the severity and incidence of doxorubicin-associated cardiomyopathies. The agent inhibits free radical formation, thereby limiting the cardiotoxicity of doxorubicin, but it may also decrease doxorubicin’s antitumor effects.21
MONOCLONAL ANTIBODIES
HER2/neu is a transmembrane tyrosine kinase that is overexpressed in 20% to 30% of patients with invasive BC. Trastuzumab is a humanized Mab directed against HER2/neu-positive (+) cells. It is indicated for patients with HER2-overexpressing breast, metastatic gastric, and gastroesophageal junction adenocarcinoma. Trastuzumab binds to the extracellular domain of the HER2/neu receptor, inhibiting cells that overexpress HER2/neu. The agent causes antibody-dependent cellular cytotoxicity.22-24
Trastuzumab-Related Cardiotoxicity
Early clinical trials of trastuzumab noted cases of CHF and decreases in LVEF. Cardiotoxicity was noted in 5% of trastuzumab patients on monotherapy, 13% of patients receiving combination therapy with paclitaxel, and 27% of patients receiving combination therapy with an anthracycline.22-24
Risk factors for trastuzumab-related cardiotoxicity include dyslipidemia, diabetes, age ≥50 years, BMI >30, sequential administration of ChT, diminishing LVEF prior to therapy, and prior treatment with anthracyclines (cumulative dose >300 mg/m2). Trastuzumab-associated cardiotoxicity, unlike AAC, is reversible.22-24
The greatest risk of trastuzumab-related cardiotoxicity occurs when the drug is given in combination with an anthracycline. Non–anthracycline-containing combination regimens with similar therapeutic efficacy are preferred. Cardiovascular events involving trastuzumab include reduced heart function, HTN, irregular heartbeat, CHF, MI, and death. The patient’s heart function should be monitored prior to initiation and during therapy.22-24
Trastuzumab’s long half-life, which ranges from 2 to 12 days depending upon the dosage, may contribute to a lack of improvement in a patient’s cardiac symptoms after discontinuation. Cardiomyocytes are not destroyed by trastuzumab but appear normal, with changes noted only by electron microscopy. Trastuzumab causes left ventricular systolic dysfunction (LVSD) by binding to the extracellular portion of the HER2/neu receptor. This is known as type II ChT-related cardiac dysfunction (CRCD), which is usually reversible, with a recovery period of 2 to 4 months. Type II CRCD is not dose-related and does not cause ultrastructural abnormalities. Anthracyclines induce type I myocardial damage, which is permanent and cumulative dose–related and may recur, depending upon additional cardiac stress.22-24
Trastuzumab should be withheld in patients exhibiting a ≥16% decrease in LVEF from initial levels. If LVEF is less than normal standardized levels and there is a ≥10% decrease from initial levels, therapy should be withheld. Therapy may be reinstituted within 4 to 8 weeks if LVEF returns to normal and there is no decrease from initial levels. Patients with persistent cardiomyopathies on three occasions or with an LVEF decrease lasting >8 weeks should discontinue trastuzumab.22-24
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is a HER2-targeted antibody-drug conjugate consisting of humanized Mab trastuzumab (T) linked to a maytansine derivative (DM1). It exerts the pharmacologic properties of a vinca alkaloid via the stable thioether linker MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). T-DM1 is indicated for use in HER2+ metastatic BC patients who received prior therapy with a taxane plus trastuzumab sequentially or in combination. It is dosed at 3.6 mg/kg once every 3 weeks until the disease advances or the patient experiences intolerable toxicity.25,26
T-DM1 was approved following the international phase III open-label EMILIA trial, which randomized 991 patients to either T-DM1 (n = 495) or lapatinib plus capecitabine (L-C) (n = 496). Inclusion criteria included a baseline LVEF of ≥50% assessed by MUGA or ECHO prior to enrollment. Patients with serious cardiac arrhythmias, unstable angina, MI, or symptomatic CHF were excluded. During the trial, most patients had a consistent LVEF of ≥45% (97.1% of T-DM1 patients vs. 93% of L-C patients). Three patients in each group had an LVEF ≤40% from baseline levels. Eight T-DM1 patients (1.7%) and seven L-C patients (1.6%) had an LVEF of <50% and ≥15% below baseline. At the time trial results were published, no L-C patients and one T-DM1 patient had developed grade 3 LVSD.25,26
There is no literature concerning the use of T-DM1 in patients with an LVEF of <50% prior to commencement of therapy. T-DM1 therapy should be withheld if LVEF is <40% or between 40% and 45% with a ≥10% decrease below initial levels. LVEF should be retested in 3 weeks; if there is no improvement, therapy should be permanently discontinued. Assessment of LVEF prior to initiation of T-DM1 is indicated, and LVEF should be monitored once every 3 months during therapy.25,26
ALKYLATING AGENTS
Alkylating agents bind to N7-guanine, inhibiting DNA replication and transcription. Cases of endomyocardial and pericardial fibrosis have been noted with busulfan. These cases, with an onset of 4 to 9 years post therapy, occurred in patients who received doses >600 mg.5,27
Cyclophosphamide-associated cardiotoxicity is related to the total dose administered, rather than to cumulative dosing. Patients receiving bone marrow transplants usually receive high doses of cyclophosphamide and therefore are predisposed to a higher level of drug-induced cardiotoxicity. Previous anthracycline therapy and mediastinal RT are predisposing factors that contribute to cyclophosphamide cardiotoxicity. Other cardiac events associated with cyclophosphamide therapy include pericarditis, HF, and myocarditis. The hypothesized MOA is a toxic metabolite that causes myocytic and endothelial injury. These toxic effects are usually acute and last for approximately 6 days.5,27
Ifosfamide (Ifex) has been reported to cause dose-related arrhythmias and HF. Cisplatin, a platinum agent, can also cause HTN, which may be precipitated by the drug’s dose-limiting effect of nephrotoxicity. Electrolyte imbalances such as hypokalemia and hypomagnesemia can predispose cisplatin patients to the development of cardiac arrhythmias. Other cardiac dysfunctions associated with cisplatin include increased cardiac enzymes that may indicate MI, palpitations, and chest pain. Patients receiving combination ChT with cisplatin and cyclophosphamide have developed HF. Patients treated with cisplatin for metastatic testicular cancer have developed myocardial ischemia, MI, LV hypertrophy, and HTN 10 to 20 years post therapy.5,27
Mitomycin is indicated for gastric and pancreatic cancer. When used in combination with an anthracycline, mitomycin has been associated with cardiomyopathies. These cardiomyopathies are probably related to the cumulative dose. Cardiotoxic effects may be produced by superoxide radicals, which are created when mitomycin is reduced under aerobic conditions to a semiquinone radical.5,27
FLUOROPYRIMIDINES
Fluorouracil (5-FU) cardiotoxicity, which occurs at a rate of 1% to 68%, usually manifests within 72 hours after administration. Angina is the most common symptomatic cardiac effect of 5-FU therapy. Arrhythmias, MI, cardiogenic shock, HF, and sudden death have occurred. Risk factors associated with 5-FU–related cardiotoxicity include prior mediastinal RT, past medical history of coronary artery disease (CAD), and combination therapy with cisplatin. Continuous infusion of 5-FU and doses >800 mg/m2 predispose patients to a greater rate of cardiotoxicity.5,27
Capecitabine (Xeloda) has a cardiotoxic rate ranging from 3% to 9%. Patients can develop symptoms of angina within 3 hours to 4 days after therapy initiation. The main risk factor for cardiotoxic events is a past medical history of CAD.5,27
Possible MOAs of fluoropyrimidine-induced cardiotoxicity include autoimmune response, coagulation-system activation, coronary artery thrombus, protein kinase C–mediated vasospasm, and toxicity to the heart muscle.5,27
TYROSINE KINASE INHIBITORS
Tyrosine kinase inhibitors (TKIs), unlike chemotherapy, do not kill nonmalignant cells, but are targeted to a specific receptor. TKIs are enzymes that cause the exchange of a phosphate residue from adenosine triphosphate (ATP) to tyrosine residues in other proteins (substrates). Because TKIs target only malignant cells, their toxicity profile is less severe than that of a ChT agent. There are two types of TKIs: Mabs, which target the external growth factor receptor, and small molecular entities, which target receptor and nonreceptor TKIs. Cardiotoxicity is one of the most prominent adverse effects (AEs) of TKI therapy.28
TKI-Associated Cardiotoxicity
Cardiovascular side effects of TKI therapy include QT prolongation, acute coronary syndromes, HF, and left ventricular dysfunction. The two types of toxicity associated with TKI therapy are on-target toxicity and off-target toxicity. On-target toxicity occurs when the tyrosine kinase responsible for regulating the production and/or life of the cancer cell also is deeply involved in the cardiomyocyte’s survival. When the cardiomyocyte is inhibited, cardiac dysfunction begins. Inhibition of a kinase that is not the original target of the TKI results in off-target toxicity. Off-target toxicity is associated with multitargeting and TKI nonselectivity. This type of nonselectivity causes an increase in AEs from the TKI.28
Systolic dysfunction causing HF is one of the most prominent AEs of TKI therapy. Pathways that regulate survival of normal cells, such as cardiomyocytes, also cause the pathologic survival and abnormal proliferation of malignant cells. Geriatric patients, renal patients, and patients with prior cardiovascular-associated dysfunction are predisposed to cardiac dysfunction from TKIs.28
Profiles of Individual TKIs
Imatinib: Imatinib (Gleevec) is used for all phases of Philadelphia chromosome+ (PH+) chronic myelogenous leukemia (CML) and also for gastrointestinal stromal tumors (GISTs). Imatinib inhibits Bcr-Abl tyrosine kinase and is associated with cardiac disorders such as pulmonary edema, tachycardia, palpitations, CHF, pericardial effusion, angina pectoris, MI, cardiac arrest, atrial fibrillation, and arrhythmias.29
Dasatinib: Dasatinib (Sprycel) is a TKI for newly diagnosed chronic-phase CML patients or CML patients intolerant or resistant to imatinib therapy, as well as for PH+ ALL patients resistant or intolerant to previous treatment. Patients on dasatinib therapy must be monitored for cardiac dysfunction such as QT prolongation.30
Nilotinib: Nilotinib (Tasigna) is indicated for CML. Patients have experienced QT prolongation and precipitous fatal events. These fatal events have occurred in Ph+ CML patients resistant or intolerant to therapy and are associated with ventricular repolarization. ECG should be done at baseline and 7 days after therapy initiation. ECGs should be performed regularly thereafter and after any dosage adjustments.31
Prior to therapy initiation, electrolyte disturbances such as hypokalemia and hypomagnesemia must be corrected. Patients with these electrolyte disturbances or long QT syndrome should not receive nilotinib. The use of strong CYP3A4 inhibitors and other drugs that may lengthen the QT interval should be avoided. Patients who must receive a strong CYP3A4 inhibitor should be considered for a dosage reduction and be closely monitored by a healthcare professional. Sudden death has been reported in patients with resistant or intolerant Ph+ CML who received nilotinib; ventricular-repolarization abnormalities may have contributed to this occurrence.31
Ponatinib: Ponatinib (Iclusig) is indicated for patients with CML (all phases) intolerant or resistant to previous treatment or for Ph+ ALL patients with the T315I+ mutation who are resistant to TKI therapy with a starting dose of 45 mg orally per day. Fatal MI, arterial thrombosis, and stroke have occurred. Arrhythmias, CHF (4%), CAD, angina, stroke, transient ischemic attack (TIA )(2%), HTN (67% treatment-emergent), and hypertensive crisis (<1%) are AEs of ponatinib therapy. Patients should be monitored for symptoms of arrhythmias and for signs and symptoms of CHF while on ponatinib. In a clinical study, 11% of ponatinib patients experienced arterial events and 8% of patients experienced serious arterial events. If serious arterial thrombosis develops, ponatinib should be discontinued.32
A small ponatinib study assessed patients for the development of QTc prolongation. No changes >20 ms from baseline were noted; however, owing to the small sample size, the possible development of QTc prolongation with this agent cannot be excluded. Ponatinib has a Risk Evaluation and Mitigation Strategy program that includes a new boxed warning, safety information, and dosing recommendations for prescribers. The boxed warning provides information on vascular occlusive episodes and a new warning regarding HF.32
Lapatinib: Lapatinib is used in combination with capecitabine for HER2/neu+ BC. Patients’ LVEF should be evaluated at baseline and during therapy. In a clinical trial, most decreases in LVEF (>57%) occurred during the first 12 weeks of therapy. Lapatinib should be discontinued in patients experiencing an LVEF decrease of grade 2 or greater or those with an LVEF lower than standardized. Electrolyte disturbances such as hypokalemia and/or hypomagnesemia can precipitate QT-interval prolongation. ECG monitoring may be essential in this subset of patients.33
Sunitinib: Sunitinib (Sutent) is a multitargeted TKI indicated for various malignancies, including renal cell cancer (RCC), GISTs, and pancreatic neuroendocrine tumors. Sunitinib has been associated with bradycardia and QTc-interval prolongation. The dose administered directly correlates with the drug’s effect on the QT interval. Sunitinib also induces CHF, with an incidence of up to 33.8% in RCC patients. ATP depletion resulting from ribosomal depletion and activation of an intrinsic apoptotic pathway is the MOA for sunitinib-induced cardiotoxicity, which is considered to be an off-target effect. Sunitinib patients have a greater rate of HF owing to the inhibition of vascular endothelial growth factor (VEGF) signaling.34
PROTEASOME INHIBITORS
Bortezomib, which is indicated for multiple myeloma and mantle cell lymphoma, has resulted in the development and worsening of HF. Patients with underlying cardiac dysfunction or risk factors should be closely monitored while on therapy.35
Bortezomib therapy has resulted in incidences and exacerbations of CHF. The proposed MOA for this cardiac dysfunction is possible dysregulation of the ubiquitin-proteasome system. When bortezomib inhibits the proteasome system, polyubiquitinated proteins accumulate, causing cardiac dysfunction by enabling systemic inhibition of the proteasome.35
TAXANES
Paclitaxel (Taxol) and docetaxel (Taxotere) work by promoting microtubule assembly and inhibiting microtubule disassembly. Bradycardia is the main cardiotoxic AE, with an incidence of 0.5% to 5% in paclitaxel patients and an incidence of 1.7% in docetaxel patients. Paclitaxel, which has proarrythmogenic properties, is associated with atrioventricular block, hypotension, sinus bradycardia, ventricular tachycardia, ischemia, and CHF. Paclitaxel affects the Purkinje system either directly with its chronotropic effects or indirectly through its Cremophor EL vehicle, which triggers the release of histamine and induces bradycardia.36
VINCA ALKALOIDS
Vinca alkaloids are antimicrotubule agents used primarily for lymphoma and leukemia patients. ECG changes, angina, MI, and myocardial ischemia have been noted with vinca alkaloids. Women experience more cardiotoxic effects from vinorelbine than do men. Vinca alkaloid–associated cardiotoxicity may be mediated by coronary spasm, which causes ischemia.36
ANTIANGIOGENIC AGENTS
Agents such as bevacizumab, sunitinib, sorafenib, pazopanib, ziv-alflibercept, and vandetanib have caused an approximately 50% increase in HTN in patients previously or currently receiving therapy.37-39
Bevacizumab (Avastin) is also an Mab and a VEGF inhibitor indicated for use in metastatic colorectal cancer, nonsquamous non-SCLC, glioblastoma, and metastatic RCC. There is an approximate 5% incidence of HTN in bevacizumab patients. VEGF inhibition enables nitric oxide suppression and prostacyclin activity in the endothelium is decreased, thereby causing the elevation in blood pressure (BP). Bevacizumab is noted for venous and arterial thromboembolic events (ATEs) including MI and cerebral infarction. Serious AEs and ATEs, including TIAs, angina, MI, and cerebral infarction, have occurred. Combination ChT with bevacizumab conferred an increased risk of ATE in patients aged ≥65 years. Patients who experience a severe ATE should not resume bevacizumab.37-39
Patients with uncontrolled BP are not candidates for bevacizumab therapy. BP should be monitored every 2 to 3 weeks during therapy. Antihypertensive agents commonly used to treat bevacizumab-induced HTN include calcium channel blockers and ACE inhibitors. Bevacizumab patients may also develop asymptomatic proteinuria. Patients with proteinuria exceeding 2 g within 24 hours should not receive bevacizumab therapy. Key factors in managing bevacizumab-induced HTN include patient education, home BP monitoring, and adjustment of medication as needed.37-39
MISCELLANEOUS AGENTS
Arsenic trioxide (Trisenox) is indicated for patients with relapsed or refractory acute promyelocytic leukemia (APL). A 12-lead ECG should be performed before arsenic therapy initiation. Patients may experience QT prolongation and should receive appropriate electrolyte monitoring, with potassium levels maintained at >4 meq/L and magnesium levels maintained at >1.8 mg/dL during therapy. Other cardiac AEs include torsades de pointes, sinus tachycardia, and fluid retention with pericardial and pleural effusions. Patients should be monitored following infusion therapy.36
All-trans retinoic acid (ATRA; Tretinoin), which is indicated for APL, is given orally at 45 mg/m2 per day in combination with an anthracycline plus or minus cytarabine in two equally divided doses until complete remission or for 90 days. Retinoic acid syndrome (RAS) can develop in approximately 26% of ATRA patients within the first 21 days of therapy. Pericardial and pleural effusions, hypotension, fever, and dyspnea are hallmarks of RAS. Approximately 17% of patients studied experienced decreased LVEF. Thrombosis and fatal MI have been noted in ATRA patients.36
CONCLUSION
Cardiotoxic cancer-related therapies present a management challenge for healthcare professionals. This morbid condition can be appropriately managed by properly educating health professionals and ensuring proper therapy administration. Cardiac-related imaging, biomarkers, and genetics are possible future sources of research strategies designed to mitigate the cardiotoxicity associated with cancer-related therapies.
REFERENCES
1. National Cancer Institute. Cancer treatment-related
cardiotoxicity: understanding the current state of knowledge and
developing future research priorities.
www.epi.grants.cancer.gov/workshops/cardiotoxicity. Accessed December 2,
2013.
2. National Institutes of Health. Cancer treatment related
cardiotoxicity (day 1) [videocast].
http://videocast.nih.gov/summary.asp?live=11985. Accessed December 1,
2013.
3. Gianni L, Herman EH, Lipschultz SE, et al. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. 2008;26:22:3777-3784.
4. Adriamycin (doxorubicin) data sheet. Auckland, New Zealand: Pfizer New Zealand Ltd; December 29, 2011.
5. Brana I, Tabernero J. Cardiotoxicity. Ann Oncol. 2010;21(suppl 7):vii173-vii179.
6. Keefe DL. Anthracycline-induced cardiomyopathy. Semin Oncol. 2001;28(4 suppl 12):2-7.
7. Safra T. Cardiac safety of liposomal anthracyclines. Oncologist. 2003;8(suppl 2):17-24.
8. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy
plus a monoclonal antibody against HER2 for metastatic breast cancer
that overexpresses HER2. N Engl J Med. 2001;344:783-792.
9. Harake D, Franco VI, Henkel JM, et al. Cardiotoxicity in childhood
cancer survivors: strategies for prevention and management. Future Cardiol. 2012;8:647-670.
10. Kalay N, Basar E, Ozdogru I, et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J Am Coll Cardiol. 2006;48:2258-2262.
11. Lipshultz SE, Giantris AL, Lipsitz SR, et al. Doxorubicin
administration by continuous infusion is not cardioprotective: the
Dana-Farber 91-01 Acute Lymphoblastic Leukemia protocol. J Clin Oncol. 2002;20:1677-1682.
12. Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int J Nanomedicine. 2007;2:567-583.
13. Caelyx (liposomal doxorubicin) product information. Macquarie Park, Australia: Janssen-Cilag Pty Ltd; October 2013.
14. Myocet (liposomal doxorubicin) product information. Utrecht, Netherlands: Teva Pharma BV; July 2010.
15. Gabizon AA. Liposomal anthracyclines. Hematol Oncol Clin North Am. 1994;8:431-450.
16. O’Byrne KJ, Thomas AL, Sharma RA, et al. A phase I dose
escalating study of DaunoXome, liposomal daunorubicin, in metastatic
breast cancer. Br J Cancer. 2002;87:15-20.
17. Fassas A, Buffels R, Anagnostopoulos A, et al. Safety and early
efficacy assessment of liposomal daunorubicin (DaunoXome) in adults with
refractory or relapsed acute myeloblastic leukaemia: a phase I-II
study. Br J Haematol. 2002;116:308-315.
18. Harris L, Batist G, Belt R, et al. Liposome-encapsulated
doxorubicin compared with conventional doxorubicin in a randomized
multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer. 2002;94:25-36.
19. Batist G, Ramakrishnan G, Rao CS, et al. Reduced cardiotoxicity
and preserved antitumor efficacy of liposome-encapsulated doxorubicin
and cyclophosphamide compared with conventional doxorubicin and
cyclophosphamide in a randomized, multicenter trial of metastatic breast
cancer. J Clin Oncol. 2001;19:1444-1454.
20. Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal
doxorubicin (doxil): reduced clinical cardiotoxicity in patients
reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000:11:1029-1033.
21. Seymour L, Bramwell V, Moran LA. Use of dexrazoxane as a
cardioprotectant in patients receiving doxorubicin or epirubicin
chemotherapy for the treatment of cancer. The Provincial Systemic
Treatment Disease Site Group. Cancer Prev Control. 1999;3:145-159.
22. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39-51.
23. Herceptin (trastuzumab) product information. South San Francisco, CA: Genentech, Inc; November 2013.
24. Jones AL, Barlow M, Barrett-Lee PJ, et al. Management of cardiac
health in trastuzumab-treated patients with breast cancer: updated
United Kingdom National Cancer Research Institute recommendations for
monitoring. Br J Cancer. 2009;100:684-692.
25. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783-1791.
26. Kadcyla (ado-trastuzumab emtansine) product information. South San Francisco, CA: Genentech Inc; May 2013.
27. Yeh ET, Tong AT, Lenihan DJ, et al. Cardiovascular complications
of cancer therapy: diagnosis, pathogenesis, and management. Circulation. 2004;109:3122-3131.
28. Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction
associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84-95.
29. Gleevec (imatinib) product information. East Hanover, NJ: Novartis Pharmaceuticals Corp; October 2013.
30. Sprycel (dasatinib) product information. Princeton, NJ: Bristol-Myers Squibb Co; June 2013.
31. Tasigna (nilotinib) product information. East Hanover, NJ: Novartis Pharmaceutical Corp; September 2013.
32. Iclusig (ponatinib) product information. Cambridge, MA: Ariad Pharmaceuticals, Inc. December 2012.
33. Tykerb (lapatinib) product information. Research Triangle Park, NC: GlaxoSmithKline; October 2013.
34. Sutent (sunitinib) product information. New York, NY: Pfizer Inc; August 2013.
35. Voortman J, Giaccone G. Severe reversible cardiac failure after
bortezomib treatment combined with chemotherapy in NSCLC: a case report.
BMC Cancer. 2006,6:129.
36. Aggarwal S, Kamboj J, Arora R. Chemotherapy-related cardiotoxicity. Ther Adv Cardiovasc Dis. 2013;7:87-98.
37. Avastin (bevacizumab) product information. South San Francisco, CA: Genentech, Inc; December 2013.
38. Granger JP, Alexander BT. Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol Scand. 2000;168:161-168.
39. BC Cancer Agency. Management guidelines for bevacizumab-related
side effects in patients with colorectal cancer.
www.bccancer.bc.ca/NR/rdonlyres/6D39414F-EC1A-4BE2-9ACB-6DE017C9B4C4/19258/Managementforbevacizumabsideeffects_1Dec06.pdf.
Accessed January 27, 2014.
To comment on this article, contact rdavidson@uspharmacist.com.


