US Pharm. 2024;49(1):4-8.
ABSTRACT: Myasthenia gravis is a neuromuscular autoimmune disease characterized by fluctuating motor weakness and clinical manifestations involving the ocular, bulbar, limb, and/or respiratory muscles. Acetylcholinesterase inhibitors, immunomodulatory and immunosuppressive drugs, plasmapheresis, IV immunoglobulin, and thymectomy are the main treatments used, depending on individual patients’ disease course and symptomatology. Myasthenia gravis is one of the most treatable autoimmune disorders; most patients improve over time with treatment and may go into remission or experience minimal symptoms. Depending on symptom severity and rate of change, treatment may be administered in an outpatient or inpatient setting.
Myasthenia gravis (MG) is an acquired autoimmune disease involving weakness of the skeletal muscles. This weakness is due to an antibody-mediated immunologic attack directed at proteins in the postsynaptic membrane of the neuromuscular junction (NMJ). Symptoms and signs, some of which include ptosis, diplopia, and changes in facial expressions, may not be immediately recognized as MG. The degree of muscle weakness varies greatly among patients. Physical and neurologic exams, along with lab panels and electrodiagnostics (electromyography), are necessary for accurate diagnosis. Myasthenia gravis is one of the most treatable autoimmune disorders, and most patients living with MG and receiving MG therapy can achieve normal function.1-3
TREATMENT OVERVIEW
Four main therapies are used for MG: acetylcholinesterase (AChE) inhibitors; glucocorticoids, nonsteroidal immunosuppressive agents, and immunomodulatory agents; plasmapheresis and IV immunoglobulin (IVIG); and surgery (i.e., thymectomy). These treatments will be discussed in detail later in this article. The time to clinical effect varies considerably by type of therapy. This factor, in addition to the pace and severity of the disease, plays a key role in choosing the appropriate treatment for each patient.1-3
The goal of MG therapy is to reduce patients’ symptoms while minimizing the medications’ side effects. Although MG is a chronic disease, many patients achieve sustained symptom remission and full functional capacity. Drug response is determined by the improvement in clinical symptoms and neurologic deficits noted on examination. Baseline neurologic function and deficits should be documented at the start of treatment and monitored for change over time as therapies are added or tapered.1
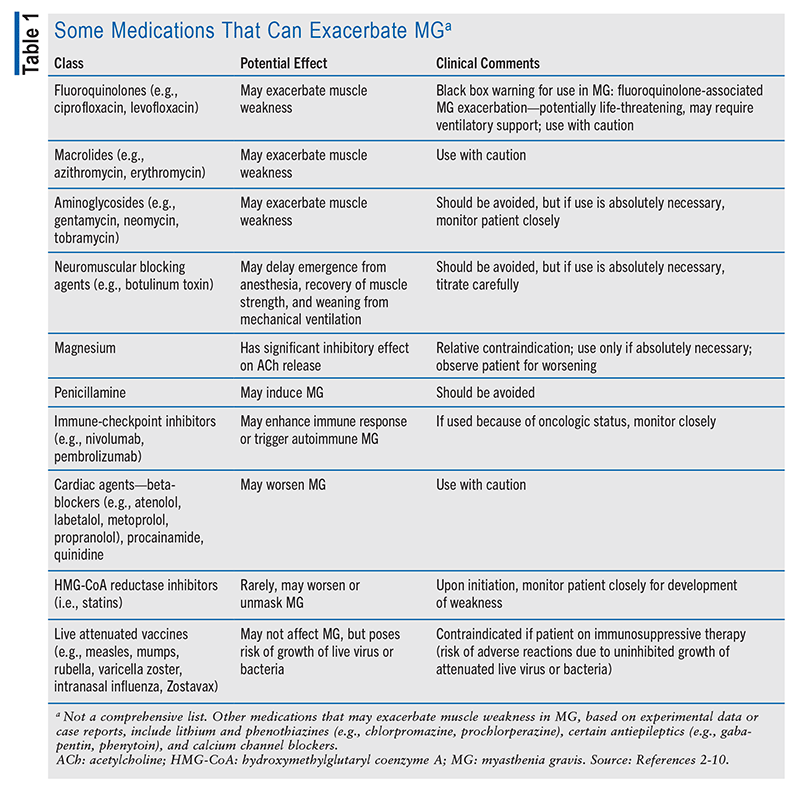
In generalized MG, a good rule of thumb is to assume that any medication can exacerbate MG and to watch for disease worsening following the introduction of a new medication. Determination of whether the association with clinical worsening is coincidental or causal may require withdrawal from the medication and a rechallenge. TABLE 1 lists some of the medications that can exacerbate MG. In addition to those listed, all drugs that are respiratory depressants (e.g., benzodiazepines, opioids, sedatives) should be used with caution in patients with diagnosed MG.2-10
SYMPTOMATIC TREATMENT
Initial therapy for most patients with mild-to-moderate MG is an oral AChE inhibitor, usually pyridostigmine. Peripherally acting AChE inhibitors are used to temporarily alleviate muscle weakness in patients with MG.11
Mechanism of Action and Response
AChE inhibitors work by reversibly inhibiting the action of AChE, preventing the breakdown of acetylcholine (ACh), which increases the amount of ACh available at the NMJ to bind to postsynaptic ACh receptors (AChRs). As a result, the effect of ACh is prolonged, leading to variable improvement in strength in patients with MG. Pyridostigmine use results in noticeable improvement in some patients and little or no improvement in others.12,13
Patients with muscle-specific tyrosine kinase (MuSK)–positive MG respond poorly to AChE inhibitors. Compared with the more common form of MG, which has antibodies against AChRs, MuSK-positive MG affects mainly the bulbar and respiratory muscles and involves more frequent, severe myasthenic crises. Therapy is usually less effective, necessitating the use of prolonged high doses of steroids and other immunosuppressants to control symptoms.12,13
With pyridostigmine use, symptom improvement may be mixed. For example, there may be resolution of weakness in one area but persistence in another. In general, limb and bulbar symptoms (dysphagia, fatigable chewing, dysarthria) respond better to AChE drugs than do ocular manifestations (ptosis, diplopia). AChE inhibitor therapy addresses symptoms only and is often insufficient in generalized MG.12,13
Dosing and Titration
Pyridostigmine has a rapid onset of action (15-30 minutes) with peak action at approximately 2 hours, and its effects last for 3 to 4 hours. Despite its short duration of action, dosing every 6 hours or three times daily may be effective in some patients; others, however, require a dose every 3 hours to maintain symptom benefit. For adults and older adolescents, a common starting dosage of pyridostigmine is 30 mg three times daily for 2 to 3 days to assess the cholinergic side effects. In patients with excessive cholinergic side effects, sometimes an agent such as oral glycopyrrolate 1 mg is added to each pyridostigmine dose to block the effects. In those who tolerate pyridostigmine well with or without anticholinergics, the dose may be increased in 30-mg increments every 2 to 3 days until a favorable therapeutic effect is achieved or further increase is limited by side effects. The maximum dosage is usually 120 mg every 4 hours during waking hours. Most adults require a total daily dosage of up to 960 mg, divided into four to eight doses, with meals. For children and younger adolescents, the initial dosage is 0.5 to 1 mg/kg every 4 to 6 hours with meals; the dose can be titrated up slowly based on therapeutic response and side effects, to a maximum daily dosage of 7 mg/kg per 24 hours, divided into five to six doses.14-18
Dosage forms for pyridostigmine include scored tablets (also available in extended-release form), a liquid formulation, and an IV preparation that may be used when a patient cannot take an oral form (generally during life-threatening neuromuscular respiratory failure, known as myasthenic crisis). As indicated above, no single pyridostigmine dosing schedule fits all patients. Most adults who respond to therapy are taking a dosage in the range of 60 to 90 mg every 4 to 6 hours during waking hours; however, some require as much as 120 mg every 3 to 4 hours during waking hours. Doses higher than this are rarely beneficial and are usually limited by cholinergic side effects. When a patient has significant persistent weakness despite sufficient doses of pyridostigmine or has side effects precluding effective dosing, immunotherapy is generally warranted.14-19
The drug regimen must be carefully individualized to achieve symptom relief and reduce cholinergic side effects. For example, many patients have significant symptoms only in the evening. In this case, an adult might be able to take the first pyridostigmine dose of the day—60 mg—at lunchtime, followed by 90 mg 4 hours and 8 hours later. Those who have trouble chewing or mild dysphagia might benefit from taking a dose 30 minutes before a meal. There are numerous potential combinations, and what works best for individual patients is based on symptom severity, response to pyridostigmine, and toleration of side effects. The use of medications that alleviate some of pyridostigmine’s cholinergic side effects may also be helpful. Extended-release pyridostigmine may be used as a bedtime dose in patients with weakness upon awakening; however, as most patients do better after a night’s sleep, it may be just as effective for those with mild weakness upon awakening to take a standard pyridostigmine dose at that time. Because of its delayed absorption and variable release, however, the use of pyridostigmine has generally been limited.14-19
Cholinergic Side Effects
Adverse effects of pyridostigmine are mostly due to the cholinergic-overstimulation properties of the drug. These cholinergic effects can be dose-limiting in many patients. The most bothersome muscarinic side effects are abdominal cramping and diarrhea; others include increased salivation and bronchial secretions, nausea, sweating, and bradycardia. Nicotinic side effects are also frequent, and these include fasciculations and muscle cramping; however, these are usually less troublesome than the gastrointestinal (GI) effects.14,15
A potential major side effect of excessive intake of an AChE inhibitor is weakness, which can be difficult to distinguish from worsening MG. This paradoxical weakening from AChE inhibitors is known as cholinergic crisis. However, cholinergic crisis occurs rarely, if ever, if pyridostigmine dosing is limited to ≤120 mg every 3 hours or a total daily dosage of up to 960 mg. Cholinergic crisis is so rare that it should not be the presumed cause of increasing weakness unless the doses taken are known to significantly exceed this range. Otherwise, even in the presence of cholinergic side effects, it should be assumed that the patient’s underlying MG is worsening, and appropriate treatment should be initiated.14,15
Side-Effect Management
Taking pyridostigmine with food can help reduce GI side effects. Muscarinic side effects can be controlled in many patients with the use of oral anticholinergic drugs that have little to no effect on nicotinic receptors (i.e., do not produce increased weakness). These include the following oral agents: glycopyrrolate 1 mg, propantheline 15 mg, and hyoscyamine sulfate 0.125 mg. These anticholinergic drugs may be taken prophylactically three times daily or, alternatively, with each pyridostigmine dose.14,15 Pronounced diarrhea can be lessened by adding loperamide or diphenoxylate hydrochloride/atropine sulfate with or without other anticholinergic drugs.14,15
IMMUNOSUPPRESSIVE TREATMENT
Although some patients do well on long-term pyridostigmine alone, most patients with generalized MG require additional treatment targeting the underlying immune dysregulation. Immunotherapy is indicated for patients who remain significantly symptomatic on pyridostigmine or who become symptomatic again after a temporary response to pyridostigmine used first-line. Glucocorticoids, nonsteroidal immunosuppressive drugs, and immunomodulatory agents are used as chronic immunotherapy. Earlier initiation of corticosteroid therapy during the disease course may lead to early- and long-term remission, with 70% to 80% of patients on steroids achieving marked improvement or complete resolution of symptoms versus 10% to 20% having spontaneous remission. Oral corticosteroids have a rapid therapeutic onset, and clinical improvement may occur within 2 weeks after therapy initiation (most improvement seen in the first 4-8 weeks). Patients with generalized MG require the addition of a nonsteroidal immunotherapeutic agent (e.g., azathioprine, tacromilus, mycophenolate mofetil) for maintenance and to spare long-term glucocorticoid side effects.19-22
Exacerbations
Acute Exacerbations: Transient worsening of myasthenic symptoms can be precipitated by concurrent infection, surgery, pregnancy, childbirth, certain medications, or tapering of immunotherapeutic drugs, or it can occur spontaneously as part of the natural disease course. When an exacerbation is severe, the patient is at risk for myasthenic crisis. Severe bulbar weakness that produces dysphagia and aspiration often complicates the respiratory failure.3,11,14
Less-Severe Exacerbations: Patients with MG frequently have worsening symptoms that are not severe enough to be considered a myasthenic crisis. As with myasthenic crisis, the initial focus of treatment is to address any external causes of the exacerbation—for example, treating a concomitant infection or stopping a medication that may worsen MG (TABLE 1). Beyond external factors, treatment should be individualized based on the rate of neurologic decline; the presence or absence of dysphagia, dyspnea, and any other major functional limitations; and the rate of onset of the various therapies. Some therapies potentially work within hours (pyridostigmine), days (IVIG, plasma exchange), or weeks (glucocorticoids). Depending on the symptom severity and rate of change, treatment may be administered in an outpatient or an inpatient care setting.3,6-8,11
IMMUNOMODULATING THERAPY
Therapeutic plasmapheresis (plasma exchange) and IVIG are immunomodulating therapies for MG. They start to work quickly (7-10 days after treatment initiation), but the treatment effects are short-lived (28-60 days). Based on limited direct comparisons, they have similar efficacy for MG, and selection of an agent is based primarily on factors related to clinician preference and patient convenience.21
Plasmapheresis
The efficacy of plasmapheresis for MG is through direct removal of pathogenic autoantibodies and complement pathway components and changes in the cytokine profile such as increased level of interleukin-10. The beneficial clinical effect of plasmapheresis is usually seen within days, but the clinical efficacy typically lasts only 3 to 6 weeks. In addition, AChR antibody levels rebound within weeks if no concurrent immunotherapy (e.g., glucocorticoids) is used. Plasmapheresis is an established treatment for seriously ill patients in myasthenic crisis. However, it is not a useful long-term treatment because the need for repeated exchanges of plasma often leads to problems with central venous access.19,22-24
IVIG
IVIG is pooled immunoglobulin obtained from thousands of donors. Its mechanism of action in MG is uncertain. As with plasmapheresis, IVIG’s efficacy typically occurs in <1 week and lasts 3 to 6 weeks. IVIG is used in the same setting as plasmapheresis to quickly reverse a severe and life-threatening exacerbation of myasthenic symptoms. IVIG also offers an alternative to plasmapheresis or multiple immunotherapeutic agents in selected patients with refractory MG; may be used as a preoperative treatment before thymectomy; or can serve as a bridge to slower-acting immunotherapies.25-31
The total dose of IVIG is 2 g/kg, usually divided over 3 to 5 days, with the most commonly used maintenance dosage (0.4 g/kg) given as a single dose every 3 to 6 weeks. In patients with renal disease or congestive heart failure and in elderly patients, it is recommended to spread out the dose over more days.19
The side effects of IVIG, including headache, chills, dizziness, and fluid retention, typically are mild and are related to the infusion rate. Uncommon complications include aseptic meningitis, acute renal failure, thrombotic events, and anaphylaxis. The acute nephrotoxicity that develops in some patients appears to be related to the high sucrose content of some IVIG preparations, and the risk increases with underlying renal insufficiency. Anaphylaxis has been associated with immunoglobulin A deficiency; however, it is rarely seen in patients treated for autoimmune neuromuscular diseases. It is also important to consider the risk of thrombotic events associated with IVIG use, including myocardial infarction, stroke, and pulmonary embolism.19,32-36
Biological Therapy
Efgartigimod alfa (monoclonal antibody that binds to Fc receptor), ravulizumab (complement C5 inhibitor), rozanolixizumab (monoclonal antibody that binds to Fc receptor), and zilucoplan (complement C5 inhibitor approved in October 2023) are biological agents used as chronic immunotherapy in patients with AChR antibody-positive generalized MG. The time to onset for these agents is a couple of weeks, so they may also be used as bridge therapy to slower-acting immunotherapies for patients in whom it is especially desirable to avoid or minimize glucocorticoid use.37
MG SUBTYPES AND SPECIAL POPULATIONS
There are important differences between MG subtypes, and additionally some patient populations require some differences in treatment. These individualized treatment approaches are briefly discussed below.
Ocular MG: The components of treatment for ocular MG are the same as for generalized MG. However, differences in symptomatology, disability, and prognosis necessitate some differences in the treatment approach for patients with ocular MG.3,11,15
MuSK-Positive or LRP4-Positive MG: Research increasingly shows that MuSK-positive MG requires important differences in treatment compared with AChR antibody-positive and seronegative MG. Most patients with MuSK-positive disease respond poorly to anticholinesterase agents as well as to thymectomy. Patients with LRP4 (LDL receptor-related protein 4)–positive MG may respond to immunotherapy, but the efficacy of thymectomy has not been established.3,11,15
Children: As with adults, treatment in children with MG should be individualized based on the severity and pace of the disease. Pyridostigmine is first-line therapy. If anticholinesterase medications do not suffice, plasmapheresis or IVIG may be used, but the effects are not long-lasting. Glucocorticoids are generally limited to severe disease that is unresponsive to these interventions. It is important to note that the safety risks associated with this drug class—including bone growth, which increases the risk of adult osteoporosis—are especially concerning when these medications are used chronically in children. Azathioprine, mycophenolate mofetil, and cyclosporine are used in juvenile MG, but concerns about serious adverse effects, including impaired fertility and late development of malignancy, are of even greater concern than in adults. The clinical outcome of childhood MG varies by age at onset, race, and sex.3,11,15-17,38-42
Pregnancy and Neonates: Pregnancy has a variable effect on the course of MG. It does not worsen the long-term outcome of MG, but MG may become more severe during pregnancy. The first trimester and the first month postpartum are the periods of highest risk of exacerbation. Transplacental passage of anti-AChR antibodies leads to transient neonatal MG in 10% to 20% of infants born to myasthenic mothers.3,11,15-17,38-42
CONCLUSION
Myasthenia gravis is one of the most treatable autoimmune diseases. Although some myasthenic patients have spontaneous remission or respond to treatment with AChE inhibitors, most need corticosteroids and/or corticosteroid-sparing drugs. A small but significant proportion of MG patients remain refractory, lack tolerance, or develop side effects to corticosteroids and immunosuppressants. The resulting unmet need for targeted immunomodulatory drugs has resulted in an ongoing campaign to develop safer, more effective treatments for MG. The recent development of biologics, which have a more targeted mechanism of action and more favorable side-effect profile, may change the treatment algorithm for MG in the future.
REFERENCES
1. Sanders DB, Burns TM, Cutter GR, et al. Does change in acetylcholine receptor antibody level correlate with clinical change in myasthenia gravis? Muscle Nerve. 2014;49:483-486.
2. Sheikh S, Alvi U, Soliven B, Rezania K. Drugs that induce or cause deterioration of myasthenia gravis: an update. J Clin Med. 2021;10:1537.
3. Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96:114-122.
4. Komal Kumar RN, Patil SA, Taly AB, et al. Effect of d-penicillamine on neuromuscular junction in patients with Wilson disease. Neurology. 2004;63:935-936.
5. Kuncl RW, Pestronk A, Drachman DB, Rechthand E. The pathophysiology of penicillamine-induced myasthenia gravis. Ann Neurol. 1986;20:740-744.
6. Zhu J, Li Y. Myasthenia gravis exacerbation associated with pembrolizumab. Muscle Nerve. 2016;54:506-507.
7. Lau KH, Kumar A, Yang IH, Nowak RJ. Exacerbation of myasthenia gravis in a patient with melanoma treated with pembrolizumab. Muscle Nerve. 2016;54:157-161.
8. Cartwright MS, Jeffery DR, Nuss GR, Donofrio PD. Statin-associated exacerbation of myasthenia gravis. Neurology. 2004;63:2188.
9. Purvin V, Kawasaki A, Smith KH, Kesler A. Statin-associated myasthenia gravis: report of 4 cases and review of the literature. Medicine (Baltimore). 2006;85:82-85.
10. Oh SJ, Dhall R, Young A, et al. Statins may aggravate myasthenia gravis. Muscle Nerve. 2008;38:1101-1107.
11. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87:419-425.
12. Punga AR, Stålberg E. Acetylcholinesterase inhibitors in MG: to be or not to be? Muscle Nerve. 2009;39:724-728.
13. Liu GT, Volpe NJ, Galetta SL. Eyelid and facial nerve disorders. In: Liu GT, Volpe NJ, Galetta SL, eds. Neuro-Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders; 2001:496.
14. Farrugia ME, Goodfellow JA. A practical approach to managing patients with myasthenia gravis—opinions and a review of the literature. Front Neurol. 2020;11:604.
15. Farmakidis C, Pasnoor M, Demachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36:311-337.
16. Ionita CM, Acsadi G. Management of juvenile myasthenia gravis. Pediatr Neurol. 2013;48:95-104.
17. Chiang LM, Darras BT, Kang PB. Juvenile myasthenia gravis. Muscle Nerve. 2009;39:423-431.
18. O’Connell K, Ramdas S, Palace J. Management of juvenile myasthenia gravis. Front Neurol. 2020;11:743.
19. Alhaidar MK, Abumurad S, Soliven B, Rezania K. Current treatment of myasthenia gravis. J Clin Med. 2022;11:1597.
20. Evoli A, Batocchi AP, Palmisani MT, et al. Long-term results of corticosteroid therapy in patients with myasthenia gravis. Eur Neurol. 1992;32:37-43.
21. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15:291-298.
22. Sghirlanzoni A, Peluchetti D, Mantegazza R, et al. Myasthenia gravis: prolonged treatment with steroids. Neurology. 1984;34:170-174.
23. Edan G, Landgraf F. Experience with intravenous immunoglobulin in myasthenia gravis: a review. J Neurol Neurosurg Psychiatry. 1994;Suppl 57:55-56.
24. Dau PC, Lindstrom JM, Cassel CK, et al. Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134-1140.
25. Newsom-Davis J, Pinching AJ, Vincent A, Wilson SG. Function of circulating antibody to acetylcholine receptor in myasthenia gravis: investigation by plasma exchange. Neurology. 1978;28:266-272.
26. Gajdos P, Chevret S, Toyka K. Plasma exchange for myasthenia gravis. Cochrane Database Syst Rev. 2002;(4):CD002275.
27. Pérez Nellar J, Domínguez AM, Llorens-Figueroa JA, et al. Estudio comparativo entre inmunoglobulina intravenosa y plasmaféresis en el perioperatorio de la miastenia gravis [A comparative study of intravenous immunoglobulin and plasmapheresis preoperatively in myasthenia] [Spanish]. Rev Neurol. 2001;33:413-416.
28. Illa I. IVIg in myasthenia gravis, Lambert Eaton myasthenic syndrome and inflammatory myopathies: current status. J Neurol. 2005;252(Suppl 1):I14-I18.
29. Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2012;(12):CD002277.
30. Patwa HS, Chaudhry V, Katzberg H, et al. Evidence-based guideline: intravenous immunoglobulin in the treatment of neuromuscular disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2012;78:1009-1015.
31. Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68:837-841.
32. Arsura E. Experience with intravenous immunoglobulin in myasthenia gravis. Clin Immunol Immunopathol. 1989;53(2 Pt 2):s170-s179.
33. Cosi V, Lombardi M, Piccolo G, Erbetta A. Treatment of myasthenia gravis with high-dose intravenous immunoglobulin. Acta Neurol Scand. 1991;84:81-84.
34. Dalakas MC. Intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: present status and practical therapeutic guidelines. Muscle Nerve. 1999;22:1479-1497.
35. Dalakas MC. The use of intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: evidence-based indications and safety profile. Pharmacol Ther. 2004;102:177-193.
36. Arsura E, Brunner NG, Namba T, Grob D. High-dose intravenous methylprednisolone in myasthenia gravis. Arch Neurol. 1985;42:1149-1153.
37. DeHart-McCoyle M, Du X. New and emerging treatments for myasthenia gravis. BMJ Med. 2023;2:e000241.
38. Lindberg C, Andersen O, Lefvert AK. Treatment of myasthenia gravis with methylprednisolone pulse: a double blind study. Acta Neurol Scand. 1998;97:370-373.
39. Lacomis D. Myasthenic crisis. Neurocrit Care. 2005;3:189.
40. Andrews PI. Autoimmune myasthenia gravis in childhood. Semin Neurol. 2004;24:101-110.
41. Tracy MM, McRae W, Millichap JG. Graded response to thymectomy in children with myasthenia gravis. J Child Neurol. 2009;24:454-459.
42. Andrews PI, Massey JM, Howard JF Jr, Sanders DB. Race, sex, and puberty influence onset, severity, and outcome in juvenile myasthenia gravis. Neurology. 1994;44:1208-1214.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



