US Pharm. 2012;37(10):29-34.
ABSTRACT: Paget’s disease (osteitis deformans), a localized disorder of the skeletal system, is characterized by increased bone turnover. The clinical presentation of Paget’s disease ranges from no symptoms to bone pain, painful skeletal deformities, or fractures. It often is discovered because of an elevated serum alkaline phosphatase (ALK-P) of unknown origin or upon radiography. The goals of treatment are to alleviate symptoms and to reduce bone turnover to prevent complications. Both calcitonin and bisphosphonates inhibit osteoclastic bone resorption and may be used to treat Paget’s disease; however, bisphosphonates are the medication of choice. Although most comparison trials of bisphosphonates report surrogate outcomes (normalization of serum ALK-P), zoledronic acid—a more potent bisphosphonate that has demonstrated efficacy and has the convenience of being administered as a single infusion—is often the preferred bisphosphonate for Paget’s disease.
Paget’s disease of bone, also known as osteitis deformans, is a localized disorder of the skeletal system that is characterized by increased bone turnover. It may involve a single bone (monostotic) or multiple bones (polyostotic).1 Approximately 34% of patients diagnosed with Paget’s disease have a monostotic lesion.2 While any skeletal site has the potential to be affected by Paget’s disease, some sites are more frequently involved, particularly the pelvis, vertebrae, femur, tibia, and skull.1 Enlarged but structurally weaker bones develop over time. Many patients have active disease for many years before presentation, since the disorder progresses at a rate of 1 cm per year in long bones.3 Over time, untreated Paget’s disease has the potential to become debilitating.
Pathophysiology
Increased osteoclastic bone resorption, greater vascularity, marrow fibrosis, and disorganized bone formation occur in Paget’s disease.4 Osteoclasts, which predominate in the early course of disease, contain more nuclei and are larger and more numerous in pagetic lesions of the bone.3,4 As the pagetic lesion matures, osteoblasts predominate and the pagetic bone is disorganized because of the faster rate of bone turnover.3,4 This leads to a reduction in mechanical strength and potentially to deformities and fractures.
Genetic factors play a role in the development of Paget’s disease. The disorder occurs more frequently in individuals of European descent aged 55 years and older.1,4 Individuals of Scandinavian, Eastern European, or Asian descent rarely develop Paget’s disease. For first-degree relatives of patients with Paget’s disease, the risk of developing the disorder is seven to 10 times that for the general population.1 Environmental factors, such as low calcium intake or childhood vitamin D deficiency, zoonotic infections, exposure to toxins, and viral infections, have been suggested as potential triggers for Paget’s disease.4 However, only the relationship between viral infections and the disease has been studied, and the results are conflicting.1
Clinical Presentation
The clinical presentation of Paget’s disease ranges from no symptoms to bone pain, painful skeletal deformities, or fractures.1 Signs and symptoms are more common with polyostotic involvement of the skeleton. Bone pain is characteristically present at rest, at night, and with use of the affected bone.4 Deformities that occur in the skull, facial bones, and extremities are more easily identifiable.1 Incomplete fissure fractures and complete fractures, which are typically transverse breaks, can be a major complication of Paget’s disease.2 Degenerative arthritis of the hips is associated with Paget’s disease occurring in the pelvis and can significantly impair mobility. This complication may result in the need for a hip replacement.
Headaches and hearing loss may be associated with Paget’s disease occurring in the skull. Hearing loss can be sensorineural, mixed, or conductive. Dental complications may result in loss of teeth if the disease occurs in the maxilla or mandibula. Neurologic complications, such as spinal cord compression, occur in up to 76% of patients and can produce considerable morbidity.1,2 Joint pain may be present when pagetic lesions affect bone segments adjacent to large joints.1 Cardiovascular complications, such as arterial calcification, are not unusual in patients diagnosed with Paget’s disease, and the increased vascularity of pagetic lesions may lead to increased cardiac output. Sarcomas complicate Paget’s disease in fewer than 1% of patients.2 Finally, hypercalcemia may develop in patients who are immobilized by severe Paget’s disease.5
Diagnosis
Since Paget’s disease often is asymptomatic, it frequently is discovered accidentally because of an elevated serum alkaline phosphatase (ALK-P) of unknown origin or upon radiography performed to evaluate abdominal symptoms or bone, pelvic, or hip pain.1,2 Ninety-five percent of untreated patients with Paget’s disease have an elevated serum ALK-P level; however, a normal concentration does not exclude the diagnosis.4 Serum ALK-P may be normal in patients with monostotic or metabolically inactive disease. Serum ALK-P elevations are reflective of an increased number of osteoblasts in the lesions.2 The level is indicative of both disease extent and disease activity.6 Serum total ALK-P levels are appropriate for following the disease, but the bone-specific isoform should be used in patients with coexisting hepatobiliary disease. Urinary deoxypyridinoline and cross-linked N-telopeptide of type I collagen, other markers of bone resorption, have not been found to confer additional benefit on serum ALK-P, and they are more costly.6
To confirm the diagnosis and determine the location and extension of Paget’s disease, radiographs and total bone scintigraphy are performed. Bone scintigraphy is more sensitive for identifying pagetic lesions; however, it has low specificity, as there are other diseases that cause bone remodeling.1 If malignancy is suspected, CT, MRI, and biopsy may be performed.
Pharmacologic Treatment
Treatment for Paget’s disease focuses on alleviating symptoms and reducing bone turnover, with a long-term goal of inducing remission to prevent progression and complications.5 Both calcitonin and bisphosphonates inhibit osteoclastic bone resorption and may be used to treat Paget’s disease. Analgesics and nonsteroidal anti-inflammatory drugs also may be employed to manage the associated symptoms.
Bisphosphonates: The treatment of choice for management of Paget’s disease is a bisphosphonate. Bisphosphonates improve bone formation by slowing bone turnover and inhibiting osteoclast activity. Randomized, controlled trials have demonstrated that bisphosphonate therapy can improve bone pain, promote healing of osteolytic lesions, decrease ALK-P levels, and restore normal bone histology.4,5 Bisphosphonates localize to hydroxyapatite in the bone. They have a long elimination from the skeleton, since they are embedded in the bone and are released during subsequent resorption through a multiple-phase process.7 Etidronate, tiludronate, pamidronate, risedronate, alendronate, and zoledronic acid are FDA approved for the treatment of Paget’s disease (TABLE 1). Although other bisphosphonates are available, this article focuses on those with an FDA-approved indication for Paget’s disease.
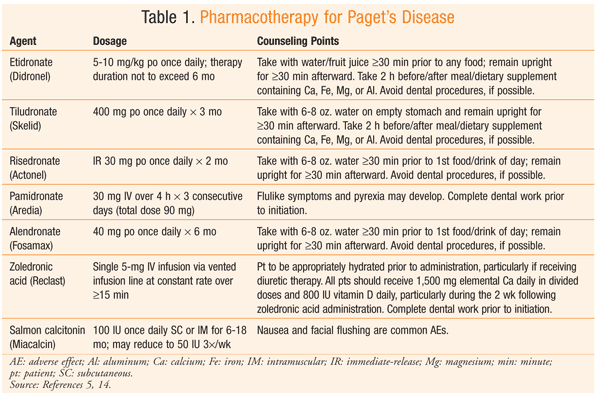
Bisphosphonates vary in their binding affinity to hydroxyapatite, with higher-affinity agents possibly leading to improved disease management.2,4 For example, zoledronic acid is about 10,000 times more potent than etidronate with regard to inhibiting osteoclastic bone resorption.4
A limited number of comparative studies of Paget’s disease have been conducted. One study compared the percentage of change in serum ALK-P between oral alendronate 40 mg daily and oral etidronate 400 mg daily for 6 months.5 At 6 months, there was a significant difference in serum ALK-P reduction between alendronate and etidronate (79% vs. 44%; P <.001). Another study compared oral risedronate 30 mg daily for 2 months with oral etidronate 400 mg daily for 6 months.8 Serum ALK-P concentrations at month 12 had normalized in 73% of risedronate-treated patients versus 15% of etidronate-treated patients (P <.001). In addition, statistically significant pain reduction was seen in the risedronate group. A single infusion of IV zoledronic acid 5 mg was compared with oral risedronate 30 mg daily for 2 months.5 Once again, normal serum ALK-P concentrations were more common with the higher-potency bisphosphonate. Approximately 89% of zoledronic acid–treated patients achieved a normal serum ALK-P level, versus 58% of risedronate-treated patients (P <.001).9 Normalization of serum ALK-P levels continued in 98% of zoledronic acid–treated patients for 2 years.5 Zoledronic acid also demonstrated a slight improvement in pain control compared with risedronate (P <.05).9
Bisphosphonates are given either IV or orally. Absorption rates of oral bisphosphonates are low (0.7%-2.5%), and absorption occurs throughout the entire gastrointestinal (GI) tract.7 All bisphosphonates have a low oral bioavailability, and bioavailability declines in the presence of food, calcium, magnesium or aluminum; therefore, oral bisphosphonates should be taken on an empty stomach with plain (nonmineral) water. Bisphosphonates are excreted unchanged in the urine through glomerular filtration. A strong correlation exists between renal excretion and renal function, so dosage adjustments can be based on creatinine clearance (CrCl).7 Bisphosphonates should not be used in patients with a CrCl under 30 to 35 mL per minute.
Pamidronate, risedronate, alendronate, and zoledronic acid are nitrogen-containing bisphosphonates; etidronate and tiludronate, the older bisphosphonates, do not contain nitrogen. Dyspepsia, esophagitis, esophageal reflux, gastritis, nausea, and other manifestations of GI pain or discomfort are the most common adverse effects (AEs) associated with oral nitrogen-containing bisphosphonates.10 Diarrhea is the most common AE associated with older, nonnitrogen-containing bisphosphonates.10 To reduce GI AEs, the patient should be instructed to refrain from lying down for 30 minutes after administration of an oral bisphosphonate. Other AEs have been tied to bisphosphonate therapy, but at a much lower incidence rate. Bisphosphonates disrupt the process of bone remodeling that repairs microdamage by reducing bone turnover; therefore, atypical femoral fractures could result.10
Osteonecrosis of the jaw (ONJ) is characterized by an area of exposed bone that will not heal, typically after dental surgery. It is a rare AE of bisphosphonates, particularly with long-term, high-dose use.4 The risk of ONJ remains extremely low with oral bisphosphonate therapy or 5 mg IV zoledronic acid once a year, the recommended dosage for the treatment of Paget’s disease.10 Ocular inflammation, mainly uveitis and scleritis, has been associated with nitrogen-containing bisphosphonates.11 Discontinuation of the bisphosphonate and a short course of corticosteroids resolve the inflammation.10 Musculoskeletal pain from bisphosphonate use has been suggested, but temporary or permanent discontinuation of the drug has provided relief.10
To avoid hypocalcemia, dietary calcium and vitamin D deficiencies must be corrected prior to bisphosphonate administration.4 In addition, patients should obtain 1,000 to 1,500 mg of calcium and 400 to 800 IU of vitamin D daily through diet or supplementation, as new bone formation occurs during the process of repairing pagetic bone.5 Calcium and vitamin D supplementation is especially important in patients receiving zoledronic acid.
Calcitonin: Calcitonin has a shorter duration of action and weaker antiresorptive effects than bisphosphonates; therefore, it is less frequently used in the treatment of Paget’s disease.4 If bisphosphonate therapy is contraindicated, salmon calcitonin is an approved alternative. Salmon calcitonin rapidly inhibits osteoclast activity and reduces levels of biochemical markers of bone turnover by up to 50% over 3 to 6 months. Lesions have been shown to resolve upon radiologic examination in the case of therapy administered for more than 1 year.2 Nausea and flushing are considered the most common AEs.
Surgery
Surgical procedures such as joint replacement, fracture fixation, and correction of bone deformity or spinal stenosis may be necessary to manage complications related to Paget’s disease.4 If joint pain is unresponsive to medical therapy, total joint arthroplasty is an option for pain relief.6 The hip is the most common site for joint arthroplasty. Pain may be diminished with tibial osteotomy to realign the knee.6 A neurosurgery consultation may be necessary for patients with spinal stenosis.
Treatment Recommendations
Patients diagnosed with Paget’s disease who have bone pain, articular or neurogenic pain, hypercalcemia due to immobilization, or hearing loss (in the case of cranial involvement) should receive pharmacotherapy.12 To prevent excessive hemorrhage in patients with Paget’s disease who are undergoing elective orthopedic surgery involving a pagetic bone, drug therapy should be given for 2 to 3 months prior to surgery to reduce the bone’s vascularity. Treatment should be considered to prevent the progression of skeletal deformities, especially in patients with a long life expectancy.2 Early treatment may be considered in asymptomatic Paget’s disease patients to reverse osteolytic lesions and reduce skeletal deformity, arthritis, and neurologic complications; however, evidence is lacking regarding the ability of pharmacotherapy to prevent complications.2,4
Follow-up for Paget’s disease includes radiographs of the lesions at 1 year and serum ALK-P activity every 3 months during the first year.2 If drug treatment is used, serum ALK-P activity should be measured every 3 to 12 months, depending upon which agent is used. These levels typically decrease within a few months of therapy.6 If serum ALK-P activity rises at least 25% and/or bone pain recurs, retreatment should be considered.2
Counseling
Counseling points regarding bisphosphonates are highly specific (TABLE 1). Good oral hygiene is important throughout bisphosphonate treatment, and patients should be reminded to inform their dentist that they are taking a bisphosphonate, especially prior to a procedure. Prevention of falls also should be discussed with patients to reduce their risk of fractures. Weight control may reduce weight-bearing pain in the extremities.6 Hypercalcemia can occur with complete immobilization, so a tolerable amount of activity should be encouraged.6 Adherence may be affected by the strict administration guidelines for oral bisphosphonates, so the patient’s active participation in care and the encouragement of adherence by multiple providers are important.
Conclusion
Although their mortality rates do not differ from those in the general population, individuals with Paget’s disease may experience many complications, which can greatly impact their quality of life.5 The increased bone remodeling resulting from osteoclast-mediated bone resorption followed by disorganized osteoblast-mediated bone deposition can cause pain, fractures, and skeletal deformities.13 Bisphosphonates improve bone pain, improve bone formation, and reduce serum ALK-P; therefore, they are the preferred treatment modality. Although most comparison trials of bisphosphonates report surrogate outcomes (normalization of serum ALK-P), no trials have effectively evaluated endpoints such as prevention of joint replacements or deafness.3 Therefore, zoledronic acid, with its demonstrated efficacy and convenient single-infusion administration, is often the bisphosphonate of choice. This intervention shows promise of a sustained improvement in quality of life for patients with Paget’s disease.
REFERENCES
1. Falchetti A, Masi L, Brandi ML. Paget’s disease of bone: there’s
more than the affected skeletal—a clinical review and suggestions for
the clinical practice. Curr Opin Rheumatol. 2010;22:410-423.
2. Singer F. Paget disease: when to treat and when not to treat. Nat Rev Rheumatol. 2009;5:483-489.
3. Reid IR, Hosking DJ. Bisphosphonates in Paget’s disease. Bone. 2011;49:89-94.
4. Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget’s disease of bone. Lancet. 2008;372:155-163.
5. Silverman SL. Paget disease of bone: therapeutic options. J Clin Rheumatol. 2008;14:299-305.
6. Whyte MP. Paget’s disease of bone. N Engl J Med. 2006;355:593-600.
7. Cremers S, Papapoulos S. Pharmacology of bisphosphonates. Bone. 2011;49:42-49.
8. Miller PD, Brown JP, Siris ES, et al. A randomized, double-blind
comparison of risedronate and etidronate in the treatment of Paget’s
disease of bone. Am J Med. 1999;106:513-520.
9. Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med. 2005;353:898-908.
10. Pazianas M, Abrahamsen B. Safety of bisphosphonates. Bone. 2011;49:103-110.
11. Etminan M, Forooghian F, Maberley D. Inflammatory ocular adverse
events with the use of oral bisphosphonates: a retrospective cohort
study. CMAJ. 2012;184:E431-E434.
12. Favus MJ, Vokes TJ. Paget’s disease of bone. In: Longo DL, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Professional; 2012.
13. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032-1045.
14. Micromedex Healthcare Series [Internet database]. Greenwood Village, CO: Thomson Reuters; 2012.
To comment on this article, contact rdavidson@uspharmacist.com.



