US Pharm. 2021:46(7):HS-8-HS-12.
ABSTRACT: Idiopathic pulmonary fibrosis (IPF) is an irreversible, chronic, and progressive pulmonary disorder characterized by thickening and scarring of the lung tissue. The disease course of IPF can be unpredictable, but it is associated with a poor prognosis with an average life expectancy of 2 to 4 years after diagnosis. Common symptoms include shortness of breath and a persistent dry, hacking cough. Although the cause of IPF is unknown, risk factors for the development of IPF include smoking, certain genetic mutations, and possibly gastroesophageal reflux disease. There is no cure. Management of IPF is multifaceted and may include the use of the antifibrotic agents nintedanib and pirfenidone, smoking cessation, and supportive care, including oxygen therapy, pulmonary rehabilitation, and in some cases, lung transplantation. Pharmacists can educate patients about treatments for IPF and make clinical recommendations when warranted that are tailored to patient-specific needs.
Idiopathic pulmonary fibrosis (IPF) is defined as an unpredictable, irreversible, progressive, and fatal lung disease developing from an unknown cause that affects the tissue surrounding the air sacs or alveoli.1 IPF has a negative effect on patient quality of life and productivity, and it is associated with high rates of mortality.1 The disease is characterized by fibrosis, permanent scarring and thickening of the lung tissue.1-3 As the disease advances, it becomes more difficult to breathe and the lungs are unable to expand and contract effectively. This results in irreversible damage to the lungs and a loss of the lung tissues’ capacity to obtain and transport oxygen to the body.2,3 In the United States, approximately 100,000 individuals are affected by IPF, and about 30,000 to 40,000 new cases are diagnosed annually.4,5 The prognosis for IPF is very poor; the average survival is 2 to 4 years after diagnosis. An estimated 50% of patients die within 2 years of diagnosis, and about 81% die within 5 years of diagnosis.3,6-8 The majority of patients with IPF first start observing symptoms between the ages of 50 and 70 years; it is more prevalent among men, but the number of cases of IPF in women is escalating.8
Risk Factors for IPF
In the past decade, the prevalence of IPF appears to be increasing, and although considered idiopathic in nature, research efforts have linked the disorder to various genetic and environmental risk factors.6,9,10 Risk factors may be classified as intrinsic, extrinsic, and those related to other comorbidities. There is growing clinical data to support the role of intrinsic risk factors such as genetics, aging, gender, lung microbiome, comorbidities such as gastroesophageal reflux, obstructive sleep apnea, diabetes mellitus, herpes virus infection, and hepatitis C infections, and extrinsic risk factors such as cigarette smoking, environmental exposures, and air pollution in the development of IPF.10 Moreover, these risk factors may independently boost susceptibility for IPF or act in a synergistic manner to contribute to heightened risk for development of this disease.10
Signs, Symptoms, and Diagnosis
IPF is challenging to diagnose; an estimated 50% of cases are initially misdiagnosed.10-14 The clinical presentation associated with IPF is nonspecific, and symptoms often resemble other pulmonary and cardiac conditions.10,12 According to the guidelines for IPF issued jointly by several international associations, including the American Thoracic Society, the diagnosis of IPF warrants a multidisciplinary approach and entails obtaining a detailed clinical history, thorough examination, and exclusion of other identified causes of interstitial lung disease (i.e., exposure to occupational and environmental toxins, connective tissue disease, and drug toxicity).15,16 A diagnosis of IPF may be confirmed based upon the results of several specialized tests that may include:
• Pulmonary function tests such as a spirometry (measures forced vital capacity [FVC], the amount of air that an individual can forcibly exhale after taking a deep breath).
• The 6-minute walk test (measures distance walked
and oxygen saturation during exercise; heart rate; and blood pressure).
• Lung biopsy, bronchoscopy, chest x-ray, or high-resolution CT scans.10,14-17
There are no formal staging guidelines for IPF; the degree of IPF severity is often established based on spirometry scores as indicated in TABLE 1.17
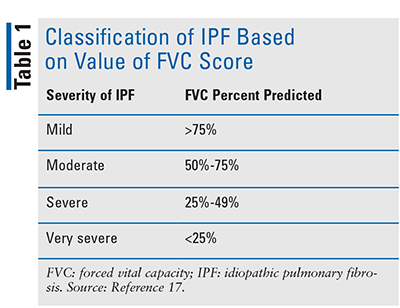
According to the IPF Foundation, estimated percentages of IPF patients who remain undiagnosed based on the severity of the disease are 52%, 33%, and 17% for mild, moderate, and severe cases of IPF, respectively.13 The most common clinical presentation is a gradual increase in dyspnea upon exertion and a dry, persistent, and nonproductive cough over several months (>6 months).12 Other symptoms may include fatigue, sleeping difficulties, gastrointestinal issues, anxiety, and depression.14 Some patients may present with chest discomfort and finger clubbing.10 During a physical examination, clinicians may hear bibasilar crackles (also known as “Velcro crackles” which sound comparable to separating a strip of Velcro). These Velcro crackles may not always be present or may only be audible unilaterally in early stages of the disease.10,12-14,18,19 Patients with more advanced disease may have end-inspiratory “squeaks” due to traction bronchiectasis, which is defined as a distortion of the airways secondary to mechanical traction on the bronchi from the existence of fibrosis of the surrounding lung parenchyma.12,20 Finger clubbing is usually an indication of advanced IPF and occurs in approximately 45% to 75% of patients.12 Early referral to a specialist is crucial to ensure prompt and accurate diagnosis, so that proper treatment can be initiated.12,14,18
Treatment of IPF
Since there is no cure for IPF, the key goal in treatment is to preserve patient lung function, decrease progression of the disease, and improve overall health-related quality of life for the patient.14,19 Early and accurate diagnosis of IPF is significant, as management options including supportive-care measures, use of antifibrotic medications, supplemental oxygen treatment, cough management, and pulmonary rehabilitation (which can include special exercises or breathing techniques) can help patients live with IPF.16,19,21 Some individuals may be candidates for lung transplants—IPF is the most common interstitial lung disease for referrals for lung transplantation and the second most frequent disease for which lung transplantation is performed.18 Palliative care is often part of the treatment plan for IPF during all stages of the disease, and patients with advanced IPF may benefit from hospice care if chosen by the patient and family members or caregivers.18
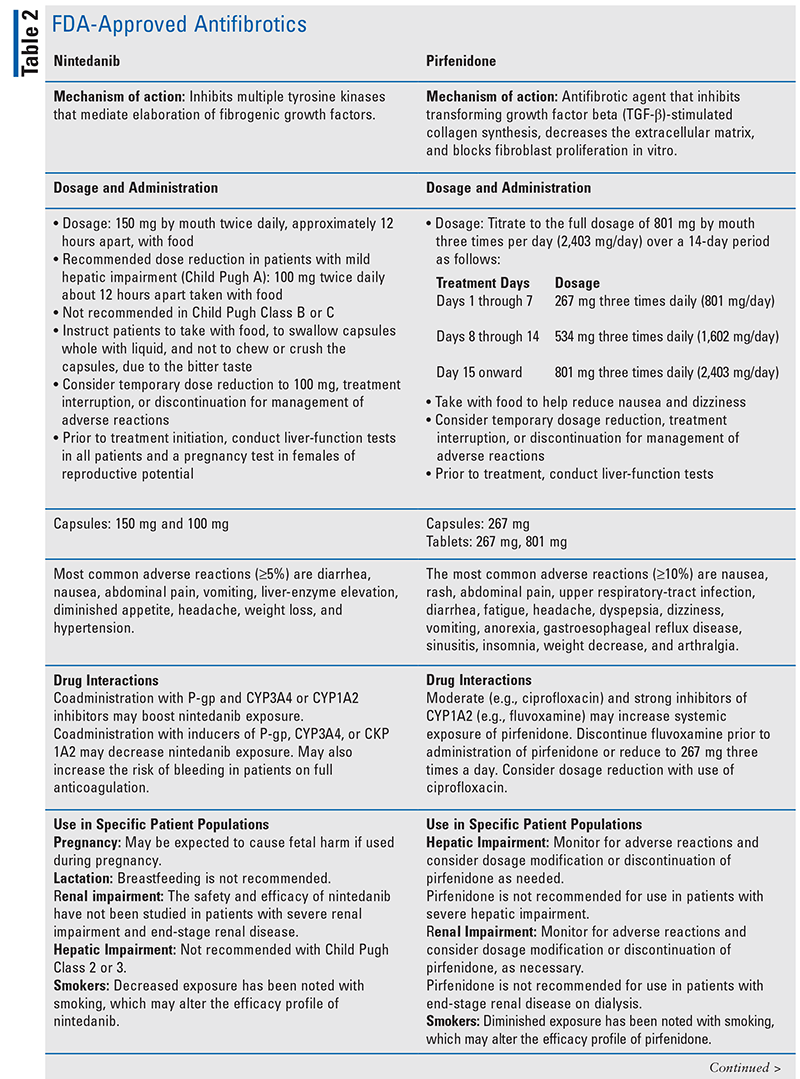
There are two approved therapies used to slow the progression of the disease.22 In 2014, the FDA approved the antifibrotics nintedanib and pirfenidone in a noteworthy breakthrough in the treatment of IPF (TABLE 2).23 The efficacy data of pirfenidone and nintedanib demonstrate the ability of both of these agents to slow down the decline of lung function in IPF patients.24,25 Results from large clinical trials have demonstrated that nintedanib and pirfenidone decrease the rate of decline in FVC by approximately 50% over 1 year of treatment in patients with IPF with mild-to-moderate impairment in lung function (FVC >50% predicted).26,27
Current research efforts are focused on the development of new drugs that target not only the deposition of extracellular matrix but also other pathways including those mediated by immunity, as well as the use of a combination of novel agents with currently available therapies.24
Nintedanib
Nintedanib is a multitargeted tyrosine kinase inhibitor that is indicated for the treatment of IPF. It acts via impeding the signal pathways of vascular endothelial growth factor receptors, platelet-derived growth factor receptors, and fibroblast growth factor receptors.25,28 By blocking these specific signal pathways, it can inhibit the progression of IPF and slow the rate of decline in pulmonary function.25,28 Reports of arterial thromboembolic events have been documented in patients while taking nintedanib; this can be of concern in patients with greater cardiovascular risk. Another constraint is the exclusion of use in pregnant and breastfeeding women.28 Based on findings from animal studies and its mechanism of action, it is thought that use of nintedanib can cause harm when administered during pregnancy.28 Bleeding events have been reported; and prescribers should assess the risks versus the benefits in those patients with known bleeding risk.28
Pirfenidone
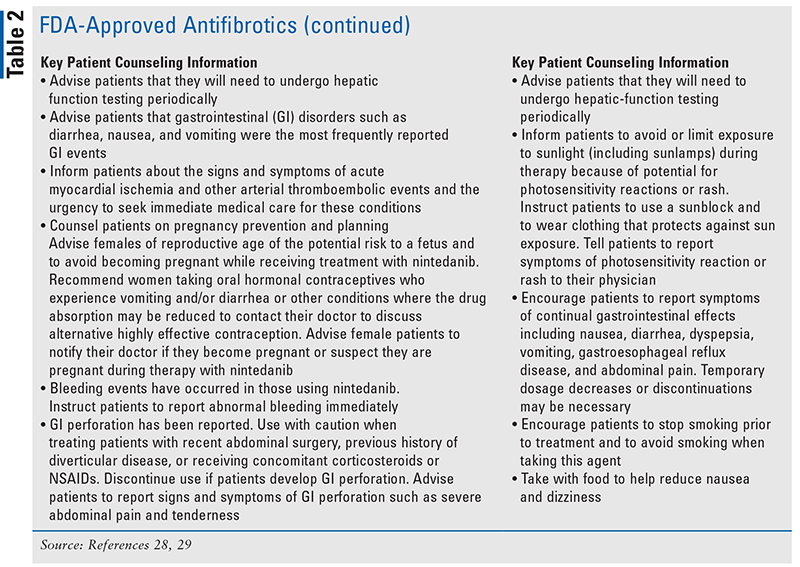
Pirfenidone is classified as a pyridone indicated for the treatment of IPF. It is considered a low-molecular-weight compound with anti-inflammatory, antioxidant, and antifibrotic effects.25,29 Pirfenidone inhibits transforming growth factor beta (TGF-β)-stimulated collagen synthesis, decreases the extracellular matrix, and blocks fibroblast proliferation in vitro.25,29 The use of pirfenidone is not recommended in those with severe hepatic impairment or in those patients with end-stage renal disease on dialysis.29 Patients should be advised to avoid exposure to sun or sunlamps due to photosensitivity and rash and be advised to wear sunscreen and protective clothing.29
Supportive Care
Supportive care is an integral component of IPF treatment and management and can include supplemental oxygen, pulmonary rehabilitation, patient education about IPF and its management and the importance of obtaining recommended vaccines, including the seasonal influenza and pneumococcal vaccinations, and palliative care throughout all stages of the disease.18,30 Patients with advanced disease may benefit from hospice care.18,30
The Role of the Pharmacist
Treatment of IPF is multifaceted, and pharmacists can be influential in recommending evidence-based pharmacologic agents when medication is warranted in IPF patients. They can also review and monitor for drug/drug interactions, potential contraindications, and potential adverse drug effects. During counseling, pharmacists can emphasize the importance of follow-up with clinicians to assess response to therapy and the significance of reporting any concerns to their prescriber. Pharmacists can augment adherence by recommending patients use medication reminders for taking and refilling medications and ensuring that patients/caregivers understand the critical nature of compliance.
Pharmacists can be instrumental in recognizing the risk factors and symptoms associated with IPF and encouraging patients exhibiting signs/symptoms or those with risk factors to seek medical evaluation. Additionally, pharmacists can be instrumental in the care of IPF patients by making clinical recommendations tailored to patient need. Pharmacists can also assist patients living with IPF by helping them to overcome the daily challenges associated with IPF, such as directing them to explore the various patient assistance/support programs, monitoring for polypharmacy, and recognizing signs of noncompliance. As patient educator and advocate, pharmacists can also encourage patients to take an active role in their treatment plan and to keep the lines of communication open with their healthcare providers. Through a collaborative, patient-centered approach to care and patient education, pharmacists are particularly qualified to enhance adherence and improve quality of life in those who suffer with IPF.
As experts in pharmacotherapy, pharmacists can provide pertinent clinical information to other healthcare professionals and patients suffering with IPF about the most recent clinical data regarding the safety and efficacy of the two FDA-approved antifibrotic agents and agents under investigation in clinical trials. Available trials for IPF can be found on the NIH Clinical Trials website: https://clinicaltrials.gov/ct2/results?cond=IPF&term=&cntry=&state=&city=&dist=.
During counseling, it is crucial to advise patients on what to expect from prescribed therapies, any precautions, and any potential adverse effects they may experience and how to manage them. Pharmacists also should inform patients that these therapies will just slow the progression of the disease and not treat IPF or reverse the damage it has caused. Prior to initiating treatment with agents such as pirfenidone and nintedanib, an in-depth conversation with the patient and family member should take place and patients should be counseled on the benefits, risks, and limitations of therapy. It is important that pharmacists counsel patients on the pharmacologic action of the selected agent, dosing, and administration and review the most common and serious adverse drug reactions. Research shows that early educational programs can help to augment patient knowledge of IPF, enabling patients and family members to better comprehend the consequences of this persistently progressive and fatal disease.31 This knowledge also allows patients and their family members to make informed choices about their health and the management options.
Pharmacists can also direct patients to helpful resources about IPF, such as Open Doors for patients taking nintedanib; and the Esbriet Inspiration Program for pirfenidone (TABLE 3, available online). The IPF Foundation provides information and supports research.
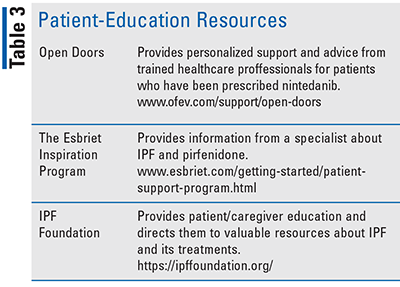
Disease-management programs are especially significant when dealing with a progressive disease like IPF, since patients may experience an overwhelming lack of psychosocial support, educational resources, and information regarding treatment options, patient drug-assistance programs, supplemental oxygen, pulmonary rehabilitation, and transplantation. They can also direct patients to the education and patient-savings resources available from the manufacturers of the antifibrotic agents nintedanib and pirfenidone.
Conclusion
The disabling and relentless symptoms of IPF, its poor prognosis, and uncertainties around the course of the disease can have an overwhelming impact on the lives of patients and their families. A collaborative effort between pharmacists, pulmonary professionals, and patients/caregivers, coupled with patient education and emphasizing the vital nature of patient adherence, is fundamental for efficaciously managing IPF, improving patient quality of life, and delaying the progression of this relentless pulmonary condition.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
REFERENCES
1. Krishna R, Chapman K, Ullah S. Idiopathic pulmonary fibrosis. Updated August 16, 2020. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. January 2021. Accessed June 15, 2021.
2. Idiopathic pulmonary fibrosis. [Internet]. Bethesda (MD): National Heart, Lung, and Blood Institute website. www.nhlbi.nih.gov/health-topics/idiopathic-pulmonary-fibrosis. Accessed June 15, 2021.
3. About Idiopathic pulmonary fibrosis. Idiopathic Pulmonary Fibrosis Foundation; 2018. https://ipffoundation.org/support-ipf-research-donate-now/. Accessed June 15, 2021.
4. Idiopathic pulmonary fibrosis. Medline Plus. Accessed April 30, 2021. https://medlineplus.gov/genetics/condition/idiopathic-pulmonary-fibrosis/.
5. Baucom M. IPF: Statistics, facts, and you. Healthline. www.healthline.com/health/managing-idiopathic-pulmonary-fibrosis/ipf-facts. Accessed May 1, 2021.
6. Sauleda J, Nunez B, Sala E, Soriano JB. Idiopathic pulmonary fibrosis: epidemiology, natural history, phenotypes. Med Sci. 2018;6(4):110.
7. Fabrellas EF, Sánchez RP, Abad CS, Samper GJ. Prognosis and follow-up of idiopathic pulmonary fibrosis. Med Sci (Basel). 2018;6(2):51.
8. American Lung Association. Pulmonary fibrosis types and causes. www.lung.org/lung-health-and-diseases/lung-disease-lookup/pulmonary-fibrosis/introduction/types-causes-and-risk-factors.html. Accessed June 15, 2021.
9. Zaman T, Lee JS. Risk factors for the development of idiopathic pulmonary fibrosis: a review. Curr Pulmonol Rep. 2018;7(4):118-125.
10. Godfrey AMK. Idiopathic pulmonary fibrosis (IPF). Practice essentials, background, pathophysiology. Medscape. Updated July 15, 2019.
https://emedicine.medscape.com/article/301226-overview#a6. Accessed April 28, 2021.
11. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941-1952.
12. King TE Jr. Clinical manifestations and diagnosis of idiopathic pulmonary fibrosis. UpToDate. Updated March 21, 2021. www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-idiopathic-pulmonary-fibrosis?search=IPF&source=search_result&selectedTitle=2~56&usage_type=default&display_rank=2#H13566478. Accessed April 28, 2021.
13. IPF. IPF Foundation website. 2020. https://ipffoundation.org/. Accessed April 28, 2021.
14. Richeldi L. Idiopathic pulmonary fibrosis: current challenges and future perspectives. Eur Respir Rev. 2013;22(128):103-105.
15. Rafii R, Juarez MM, Albertson TE, Chan AL. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis. 2013;5(1):48-73.
16. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788-824.
17. PulmonaryFibrosisMD.com. Stages of idiopathic pulmonary fibrosis. http://pulmonaryfibrosismd.com/stages-of-idiopathic-pulmonary-fibrosis/. Accessed June 1, 2021.
18. King TE Jr. Treatment of idiopathic pulmonary fibrosis. UpToDate. Updated April 2021. www.uptodate.com/contents/treatment-of-idiopathic-pulmonary-fibrosis?search=%20IPF&source=search_result&selectedTitle=1~52&usage_type=default&display_rank=1. Accessed May 1, 2021.
19. Vega-Olivo M, Criner GJ. Idiopathic pulmonary fibrosis: a guide for nurse practitioners. Nurse Pract. 2018;43(5):48-54.
20. Emmons E. What is traction bronchiectasis? Medscape. Updated September 15, 2020. www.medscape.com/answers/296961-7030/what-is-traction-bronchiectasis. Accessed June 1, 2021.
21. Cottin V, Richeldi L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur Respir Rev. 2014;23:106-110.
22. Lee J. Idiopathic pulmonary fibrosis. Merck Manual for Healthcare Professionals website. Updated September 2029. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/idiopathic-pulmonary-fibrosis. Accessed April 27, 2021.
23. Aryal S, Nathan S. An update on emerging drugs for the treatment of idiopathic pulmonary fibrosis. Expert Opin Emerg Drugs. 2018;23:2:159-172.
24. Hughes G, Toellner H, Morris H, et al. Real-world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med. 2016;5(9):78.
25. Fujimoto H, Kobayashi T, Azuma A. Idiopathic pulmonary fibrosis: treatment and prognosis. Clin Med Insights Circ Respir Pulm Med. 2015;9(1):179-185.
26. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:20832092.
27. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071-2082.
28. Ofev (nintedanib) package insert. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2019.
29. Esbriet (pirfenidone) package insert. San Francisco, CA: Genentech Inc; 2016.
30. Quinn C, Wisse A, Manns ST. Clinical course and management of idiopathic pulmonary fibrosis. Multidiscip Respir Med. 2019;14:35.
31. Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med. 2017;129:24-30.
To comment on this article, contact rdavidson@uspharmacist.com.



