US Pharm. 2021;46(9):39-44.
ABSTRACT: Myelodysplastic syndromes (MDS) are a heterogeneous group of malignant blood disorders that affect the bone marrow, ultimately resulting in bone-marrow failure, acute leukemia, and death. Treatment selection for MDS is influenced by the severity of symptoms, cytopenias, cytogenetics, prognostic category, medical fitness, and patient preferences. Although erythropoiesis-stimulating agents (ESAs) help improve anemia and reduce the transfusion burden, limited treatment options exist when patients experience treatment failure with ESAs. Recent regulatory approval of luspatercept, which targets an erythropoietic cell signaling pathway, represents a major therapeutic advance in the management of anemia in MDS patients who are refractory to ESAs.
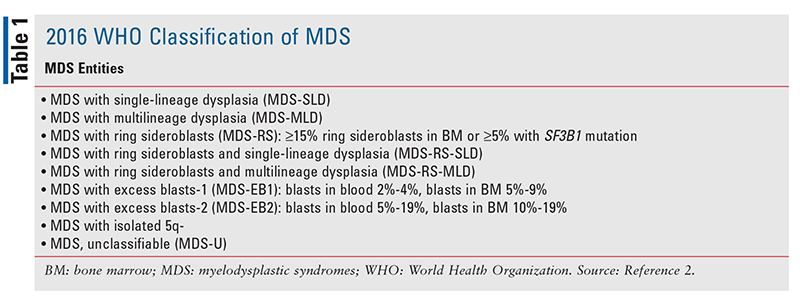
Myelodysplastic syndromes (MDS) comprise a group of hematopoietic neoplasms characterized by abnormal differentiation and cytomorphology (i.e., dysplasia) of pluripotent hematopoietic progenitor cells (i.e., stem cells) residing in the myeloid compartment of the bone marrow (BM). These abnormalities lead to ineffective hematopoiesis and to cytopenia (i.e., lower-than-normal peripheral blood cell counts) of one or more lineages of the myeloid progenitor cells that manifests as anemia, neutropenia, and/or thrombocytopenia.1 MDS ultimately result in BM failure, acute leukemia, and death. Despite their wide-ranging disease characteristics and entities (TABLE 1), these neoplasms have a common ancestral origin, being derived from the clonal expansion of somatically mutated hematopoietic progenitor cells.1,2
MDS may arise de novo (primary MDS), from chemotherapy and/or radiation for other malignancies (i.e., treatment-related), or (rarely) from environmental exposures.3 MDS are diagnosed in more than 10,000 persons in the United States annually, with an age-adjusted incidence rate of approximately 4.4 to 4.6 cases per 100,000 individuals, and the prevalence is higher in males and whites.3 Patients are at risk for symptoms related to anemia, infection, and bleeding, and they have an increased risk of progression to acute myeloid leukemia (AML).1 The median age at diagnosis is 70 years.4
This article describes the molecular pathogenesis and management of lower-risk MDS, with emphasis on the recent therapeutic advance in the treatment of anemia.
MOLECULAR PATHOGENESIS
The clinical evolution of MDS is characterized by the acquisition of multiple genetic lesions and subsequent development of clonal selection and subclonal development.5 A number of chromosomal (cytogenetic) abnormalities are observed in 50% to 60% of patients with MDS. The most frequent single cytogenetic abnormalities include deletion of the long arm of chromosome 5 (5q-), monosomy 7 (7q-), trisomy 8, and deletion of the long arm of chromosome 20 (20q-).6 Recent advances in next-generation sequencing have enabled the identification of somatic gene rearrangements and driver mutations in the pathogenesis of MDS. These genes, in their nonmutated forms, encode proteins that mediate RNA splicing, DNA methylation, chromatin modification, transcription regulation, DNA repair, and signal transduction.1,2,5 Mutations in RNA splicing factors and epigenetic regulators are the most common classes of genetic alterations in MDS, followed by mutations in transcriptional factors, tyrosine kinase signaling, and tumor suppressor TP53.5 Most of these mutated genes can also be detected in different myeloid neoplasms and are not specific to MDS. Therefore, MDS are viewed as a group of minimally to moderately developed neoplasms in the spectrum of myeloid leukemias.6
DISEASE BIOLOGY AND DIAGNOSIS
Although abnormal cytomorphology or dysplasia can be observed in neoplastic mature blood cells (red cells, white cells, and platelets) and maturing hematopoietic (erythroid, granulocytic, and megakaryocytic) cells in the BM, the neoplastic transformation of hematopoietic stem cells is the hallmark of MDS.7 A defining characteristic of MDS is the failure of the BM to successfully execute its primary physiological task of sustained hematopoiesis. This hematopoietic failure results in cytopenias, especially anemia.8 Furthermore, ineffective hematopoiesis leads to an excess of proinflammatory cytokines and to altered immune responses in T cells.9 Abnormal epigenetic changes, such as methylation of transcription promoter genes, are universal in patients with MDS, and the number of abnormal methylated genes is increased during disease progression; this makes the abnormal global methylation pattern a viable target for methyltransferase inhibitors or hypomethylating agents (HMAs) such as azacitidine or decitabine.10
MDS diagnosis generally requires at least one of the following defining events: 1) cytopenia in at least one hematopoietic lineage; 2) morphologic abnormality or dysplasia (such as dyserythropoiesis in red cells or dysgranulopoiesis in white cells) in at least 10% of nucleated cells in at least one lineage; 3) fewer than 20% blasts in the blood and BM; or 4) characteristic cytogenetic or molecular findings pertaining to MDS.2 Many factors can contribute to dysplasia of hematopoietic cells, including nutritional deficiencies (e.g., folic acid, vitamin B12, zinc, copper), iron or blood loss, toxic exposures (e.g., alcohol, heavy metals), infections (e.g., HIV, parvovirus B19), medications that can contribute to cytopenias, and heritable causes of dysplasia. Autoimmune disorders must be ruled out.11
Historically, the diagnosis of MDS was hampered by the poor reproducibility of morphologic analyses and the lack of specificity of these dysplastic changes, which made it difficult to differentiate between MDS and other clonal malignancies.5,8 The current World Health Organization classification of MDS is based on the number of cytopenic and dysplastic lineages, the percentage of blasts and ring sideroblasts, and cytogenetic findings (TABLE 2).2,11 The presence of a mutant gene, SF3B1, in the splicesome is predictive for MDS with ring sideroblasts.12 Interestingly, ring sideroblasts—so termed because of the presence of siderotic (iron) granules covering the nuclear membrane of immature red cells—are associated with an indolent disease course of MDS and prolonged survival.8
RISK STRATIFICATION AND TREATMENT OPTIONS
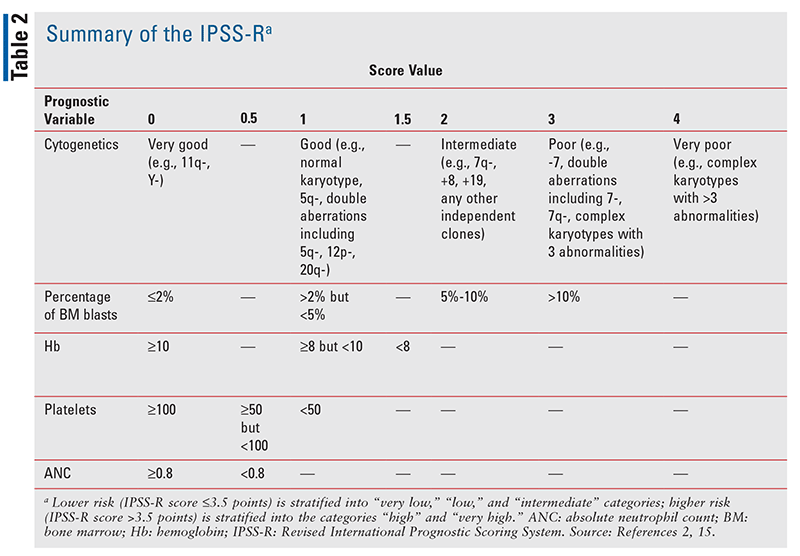
Although the disease progression of MDS is highly variable among patients, many prognostic tools have been used to predict disease risk and determine therapeutic strategies. In 1997, Greenberg and colleagues developed the International Prognostic Scoring System (IPSS) based on BM blasts (immature hematopoietic cells), cytogenetic abnormalities, and the number of cytopenias.13 This model proved to be a useful tool for predicting survival and risk of AML development in patients with MDS, with patients risk-stratified as low risk (low or intermediate-1) or high risk (intermediate-2 or high risk).13 The IPSS was revised in 2012 to incorporate percentage of BM blasts, number and severity of cytopenias, and cytogenetic findings; this new model is known as the Revised International Prognostic Scoring System (IPSS-R).14 IPSS-R categorizes MDS as either lower risk (IPSS-R score 3.5 points or below) or higher risk (IPSS-R score >3.5 points) and stratifies patients into five prognostic groups: lower risk—“very low,” “low,” and “intermediate”; higher risk—“high” and “very high” (TABLE 2).14,15
Treatment Options for Lower-Risk MDS
For lower-risk patients who are asymptomatic (i.e., no cytopenias or symptoms), active surveillance is recommended because there is no evidence that early treatment of asymptomatic lower-risk MDS improves long-term survival.11,15 Asymptomatic patients are followed periodically to evaluate the disease course and initiate supportive care if needed (e.g., antibiotics for bacterial infections, transfusions for cytopenias prior to surgery).11
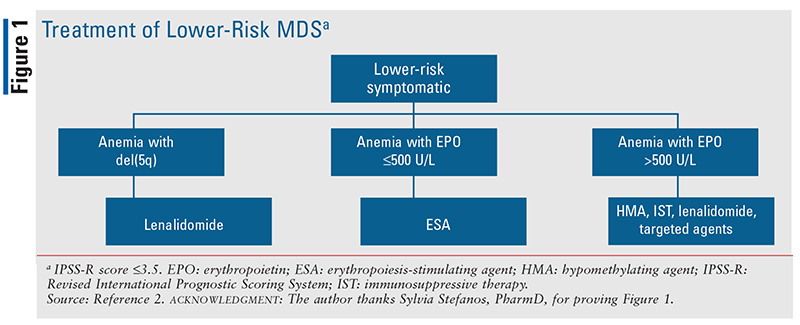
For most symptomatic patients with lower-risk MDS, lower-intensity therapy is recommended to improve symptoms, enhance quality of life, and prolong survival; therapy is not considered curative, however. The choice of a lower-intensity treatment is guided by the cell lineage(s) affected, laboratory findings (e.g., serum erythropoietin [EPO] level), cytogenetics (particularly 5q-), drug availability, and patient preferences. Examples of lower-intensity treatments include growth factors (e.g., erythropoiesis-stimulating agents [ESAs], thrombopoietin receptor agonists), HMAs, immunomodulatory drugs (IMIDs), and immunosuppressive therapy (e.g., antithymocyte globulin, cyclosporine) that aims to abrogate T-cell activity and alleviate cytopenias of MDS.2,11 See FIGURE 1.
IMIDs such as thalidomide and lenalidomide have been used to treat MDS in both prospective and retrospective nonrandomized clinical trials.16,17 These agents act to interrupt neoangiogenesis, modulate immune response, and improve the efficiency of hematopoiesis. Treatment with thalidomide as a single agent was shown to reduce or abolish transfusion dependence in a fraction of patients, but long-term treatment was significantly affected by neurotoxicity.7 The cytogenetic abnormality 5q-, which is present in 10% to 15% of MDS patients, is associated with an indolent disease course and an improved response to lenalidomide, supporting the claim that lenalidomide is selectively cytotoxic to 5q- clones and restores erythrocyte production by suppressing the myelodysplastic clone.16 The recommended dosage of lenalidomide is 10 mg by mouth daily for 21 days of a 28-day cycle. It must be noted that patients with neutropenia (<500 cells/mcL) or a low platelet count (<25,000 cells/mcL) were excluded from a lenalidomide trial in patients with 5q-.18 Therefore, treatment-related myelosuppression may still occur despite the standard 21-day administration schedule, necessitating dose adjustment or discontinuation. Dose adjustment is also warranted in renal impairment.
Despite an indolent disease course, most patients with primary MDS and 5q- eventually require RBC transfusions.19 Treatment with lenalidomide has been associated with reductions in RBC-transfusion dependence and improvements in erythroid response.18 However, additional genetic rearrangement in 5q- patients, such as TP53 mutation or 17p-, confers a poor prognosis, implying that TP53 mutation and 17p- are oncogenic drivers.20,21 Patients with these genetic abnormalities should be referred to clinical trials.20,21
Patients who have lower-risk MDS—such as those without the 5q- chromosomal abnormality—but present with moderate-to-severe anemia (hemoglobin [Hb] <10 g/dL), have a serum EPO level <500 U/L, and/or require <2 RBC transfusions per month should be considered for therapy with ESAs such as epoetin alfa (Epogen, Procrit, Retacrit) or darbepoetin (Aranesp).2,11 ESA efficacy can be further improved by the addition of a myeloid growth factor, such as filgrastim (Neupogen), tbo-filgrastim (Granix), or a biosimilar, especially in the setting of neutropenia.2,22
It should be remembered that most patients respond slowly to lower-intensity therapy such as ESAs. A meaningful improvement in Hb may require ESA treatment for 3 months or longer. Patients should remain on therapy for as long as they have clinical benefit without significant adverse effects (AEs).11 In patients who experience significant AEs (e.g., hypertension) but show evidence of response to therapy, dose reduction rather than discontinuation of therapy is warranted.11
A small subset of patients with advanced lower-risk MDS may be eligible for intensive therapies, such as intensive remission induction chemotherapy and allogeneic hematopoietic stem-cell transplantation, which can alter the natural history of MDS but are associated with substantial morbidity and mortality. These patients are typically younger and medically fit. The treatment goal is to achieve long-term disease control while minimizing treatment-related toxicities.11 Intensive therapy is also indicated in higher-risk MDS patients, who have a greater risk of progression to AML and shorter survival than lower-risk patients.11
THERAPIES FOR ANEMIA
ESAs
Anemia, a major clinical problem in MDS, is present in 85% of patients when the disease is first diagnosed.23,24 A study showed that 80% of MDS patients have an Hb level <10 g/dL at diagnosis, and most of them will become transfusion-dependent.25 Chronic anemia (Hb <10 g/dL) and RBC transfusions are associated with fatigue, risk of falls and bone fractures, impaired quality of life, increased cardiovascular risks, hepatic and endocrine dysfunctions, and iron overload, leading to shorter survival.2,23,26 Both prospective and retrospective studies have suggested that ESAs, rather than transfusions, improved the overall survival of patients with MDS.27,28 Therefore, ESAs remain the first-line agents for lower-risk MDS patients with serum EPO levels up to 500 U/L.29,30 Another favorable prognostic factor for response to ESA treatment is low or no transfusion requirement (<2 RBC transfusions per month).22 ESAs target defective erythroid development by inhibiting apoptosis and stimulating erythroid precursor cells.31 The role of the EPO level remains unclear, however.
With ESA therapy, the target Hb for lower-risk MDS patients is generally in the range of 10 g/dL to 12 g/dL or a decrease in red-cell transfusion requirements by 4 U of RBC transfusions over a period of 8 weeks.29 The ESA dosage for MDS is typically higher than that used for anemia of inflammation or chronic kidney disease because the goal is to enhance marrow responsiveness. If a response is seen at the higher dose, the dosage must be reduced to the lowest effective dose.2 In addition, fixed dosing rather than weight-based dosing is recommended in clinical practice. The feared complications of thromboembolism, seizures, and congestive heart failure are considered rare.30
A meta-analysis of studies stratified by different baseline EPO levels showed that patients with lower baseline levels (<100 U/L) had an improved response rate of 38% compared with patients with higher baseline levels (>100 U/L).31 However, it must be noted that EPO level is a continuous variable, and there is no evidence to date that MDS-associated anemia is caused by deficient endogenous production of EPO. Patients with a higher endogenous EPO level (e.g., those with refractory anemia) or a lower Hb level may still exhibit a lack of response in the BM, suggesting that early initiation of ESA may be necessary.31 On the other hand, higher doses of ESA correlated with improved erythroid response. Given the heterogeneity of the studies in this meta-analysis, the results must be interpreted with caution, and prospective, randomized studies are needed in order to elucidate these correlations.31
Moreover, it had been shown that weekly dosing of epoetin alfa 40,000 U or darbepoetin 150 mcg to 300 mcg yields approximately 60% of erythroid responses.32 Response to ESAs typically occurs within 8 weeks of treatment, although some patients may respond after 12 weeks.31 Although current literature supports daily or twice- or thrice-weekly dosing for the short-acting epoetin alfa, less-frequent dosing that enhances patient convenience may also be feasible. The long-acting form of darbepoetin may be dosed weekly or on a schedule of every 2 or 3 weeks.33,34
A baseline iron study must be performed to ensure adequate iron stores before ESA therapy is initiated. Supplemental oral or IV iron may be indicated with continued use of ESA after initial response because iron is needed for erythropoiesis. Early ESA failure (no response to ESA or relapse within 6 months) is a prognostic marker of poor disease outcome and progression to AML.32
Therapy Targeting the Transforming Growth Factor (TGF) Pathway
Despite the use of ESA for lower-risk MDS, only about one-third of unselected patients treated with ESAs achieve erythroid response. Additionally, limited treatment options exist when patients experience treatment failure with ESAs because anemia in MDS is due to ineffective erythropoiesis that cannot be corrected by exogenous ESA. These circumstances underscore the need for novel therapeutic agents.23
In MDS, key defects implicated in dyserythropoiesis and subsequent ineffective erythropoiesis are potential targets for precision medicine. Active therapeutic development includes agents targeting GATA-1 (a master regulator of erythroid differentiation and the TGF-beta superfamily signaling pathway), which is involved in the apoptosis, proliferation, differentiation, and migration of hematopoietic stem cells.35,36 TGF-beta pathway signaling is mediated by intracellular transducers known as Smads, which are encoded by genes with sequences similar to those in the S (“small”) gene of Caenorhabditis elegans and the MAD (“mothers against dpp” [decapentaplegic]) gene in Drosophila melanogaster. Smad proteins belong to a family of transcription factors with eight members—namely, Smad1 through 8—that can be subcategorized into three classes based on their structural and functional similarities.37 Under normal conditions, Smad-pathway ligands such as activins and growth-differentiation factors exert inhibitory regulatory effects on multiple phases of erythropoiesis.38 However, in certain pathologic conditions, such as MDS, this signaling pathway is dysregulated, leading to anemia.
Recently, a fusion protein (luspatercept [Reblozyl]) that binds to the TGF-beta superfamily ligands, decreases Smad2 or Smad3 signaling, and enables late-stage erythroblast differentiation has been identified as a novel therapeutic agent for patients with lower-risk MDS with ring sideroblasts. In a phase II dose-finding trial, 58 lower-risk MDS patients were treated with luspatercept.39 The erythroid response rate (ERR) was 63%, with 38% of patients achieving transfusion independence. Response was more likely among patients who harbored SF3B1 mutations compared with those who did not (77% vs. 40%, respectively). A subsequent phase III trial (MEDALIST) confirmed that, in lower-risk MDS patients with 15% or more ring sideroblasts or with 5% or more ring sideroblasts with SF3B1 mutation that were considered ESA refractory, the ERR during the first 24 weeks was 53% in the luspatercept arm (at a dose level of 1 mg/kg-1.75 mg/kg) versus 12% in the placebo arm.40 This study led to the 2020 U.S. regulatory approval of luspatercept for lower-risk MDS with ring sideroblasts and SF3B1 mutation. Common luspatercept-associated AEs included fatigue, diarrhea, asthenia, nausea, and dizziness; the incidence of these AEs decreased over time.40
Current National Comprehensive Cancer Network guidelines recommend the use of luspatercept after 2 months of ESA therapy in lower-risk MDS patients with ring sideroblasts and a serum EPO level <500 U/L.2 Luspatercept is also indicated in lower-risk MDS patients with ring sideroblasts and an EPO level >500 U/L. Nonresponders to both ESA and luspatercept may be offered immunosuppressive therapy or HMAs.2
THE PHARMACIST’S ROLE
Implementing clinical pharmacy service into routine care of hematology/oncology patients requires an interdisciplinary effort. Clinical pharmacists’ contribution to the team lies in their ability to assess the possible etiology of cytopenias due to medications or nutritional deficiencies. They can also recommend appropriate therapies to correct these reversible causes. By monitoring laboratory values, assessing IPSS-R score, triaging patient symptoms, triaging patient preferences, and developing treatment plans to support the appropriate use of ESAs, luspatercept, and other therapies, pharmacists can facilitate medication safety, ensure treatment efficacy, and reduce costs for patients with lower-risk MDS.
REFERENCES
1. Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872-1885.
2. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines)®. Myelodysplastic syndromes. Version 3.2021. www.nccn.org/professionals/physician_gls/pdf/mds.pdf. Accessed February 17, 2021.
3. National Cancer Institute. Myelodysplastic syndromes treatment (PDQ®)-health professional version. www.cancer.gov/types/myeloproliferative/hp/myelodysplastic-treatment-pdq#_291_toc. Accessed February 12, 2021.
4. Franke GN, Lückemeier P, Platzbecker U. Allogeneic stem-cell transplantation in patients with myelodysplastic syndromes and prevention of relapse. Clin Lymphoma Myeloma Leuk. 2021;21(1):1-7.
5. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): an update. Mutat Res Rev Mutat Res. 2016;769:47-62.
6. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496-2506.
7. Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943-2964.
8. Steensma DP. Historical perspectives on myelodysplastic syndromes. Leuk Res. 2012;36(12):1441-1450.
9. Kordasti SY, Afzali B, Lim Z, et al. IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br J Haematol. 2009;145(1):64-72.
10. Jiang Y, Dunbar A, Gondek LP, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113(6):1315-1325.
11. Estey EH, Sekeres MA. Overview of the treatment of myelodysplastic syndromes. UpToDate. www.uptodate.com/contents/overview-of-the-treatment-of-myelodysplastic-syndromes. Accessed January 27, 2021.
12. Malcovati L, Papaemmanuil E, Bowen DT, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118(24):6239-6246.
13. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079-2088.
14. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465.
15. Germing U, Kündgen A. Prognostic scoring systems in MDS. Leuk Res. 2012;36(12):1463-1469.
16. List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456-1465.
17. Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86-93.
18. Giagounidis A, Mufti GJ, Mittelman M, et al. Outcomes in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with isolated deletion 5q treated with lenalidomide: a subset analysis from the MDS-004 study. Eur J Haematol. 2014;93(5):429-438.
19. Giagounidis AA, Germing U, Aul C. Biological and prognostic significance of chromosome 5q deletions in myeloid malignancies. Clin Cancer Res. 2006;12(1):5-10.
20. Jädersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011;29(15):1971-1979.
21. Saft L, Li JS, Greenberg PL, et al. p53 mutant independently impacts risk: analysis of deletion 5q, lower-risk myelodysplastic syndromes (MDS) patients treated with lenalidomide (LEN) in the MDS-004 Study. Blood. 2014;124(21):414.
22. Hellström-Lindberg E, Gulbrandsen N, Lindberg G, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol. 2003;120(6):1037-1046.
23. Platzbecker U, Hofbauer LC, Ehninger G, Hölig K. The clinical, quality of life, and economic consequences of chronic anemia and transfusion support in patients with myelodysplastic syndromes. Leuk Res. 2012;36:525-536.
24. Santini V. Treatment of low-risk myelodysplastic syndromes. Hematology Am Soc Hematol Educ Program. 2016;2016(1):462-469.
25. Sanz GF, Sanz MA, Vallespi T, et al. Two progression models and a scoring system for predicting survival and planning treatment in myelodysplastic syndromes: a multivariate analysis of prognostic factors in 370 patients. Blood. 1989;74(1):395-408.
26. Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018;131(5):505-514.
27. Park S, Kelaidi C, Sapena R, et al. Early introduction of ESA in low risk MDS patients may delay the need for RBC transfusion: a retrospective analysis on 112 patients. Leuk Res. 2010;34(11):1430-1436.
28. Greenberg PL, Sun Z, Miller KB, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood. 2009;114(12):2393-2400.
29. Carraway HE, Saygin C. Therapy for lower-risk MDS. Hematology Am Soc Hematol Educ Program. 2020;2020(1):426-433.
30. Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: contemporary review and how we treat. Am J Hematol. 2016;91(1):76-89.
31. Park S, Fenaux P, Greenberg P, et al. Efficacy and safety of darbepoetin alpha in patients with myelodysplastic syndromes: a systematic review and meta-analysis. Br J Haematol. 2016;174(5):730-747.
32. Tehranchi R, Invernizzi R, Grandien A, et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood. 2005;106(1):247-253.
33. Villegas A, Arrizabalaga B, Fernández-Lago C, et al. Darbepoetin alfa for anemia in patients with low or intermediate-1 risk myelodysplastic syndromes and positive predictive factors of response. Curr Med Res Opin. 2011;27(5):951-960.
34. Kelaidi C, Beyne-Rauzy O, Braun T, et al. High response rate and improved exercise capacity and quality of life with a new regimen of darbepoetin alfa with or without filgrastim in lower-risk myelodysplastic syndromes: a phase II study by the GFM. Ann Hematol. 2013;92(5):621-631.
35. Zhao B, Liu H, Mei Y, et al. Disruption of erythroid nuclear opening and histone release in myelodysplastic syndromes. Cancer Med. 2019;8(3):1169-1174.
36. Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542-3550.
37. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783-2810.
38. Bataller A, Montalban-Bravo G, Soltysiak KA, Garcia-Manero G. The role of TGF beta in hematopoiesis and myeloid disorders. Leukemia. 2019;33(5):1076-1089.
39. Platzbecker U, Germing U, Götze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338-1347.
40. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140-151.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



