US Pharm. 2007:32(7):HS-5-HS-16.
Cystic
fibrosis (CF) is a chronic genetic disease that affects multiple bodily
systems, predominantly the lungs and gastrointestinal (GI) tract. In the
United States, approximately 30,000 children and adults have CF, and about 1
in 3,500 infants is born with CF each year.1 CF occurs most
commonly among whites. Great advances in CF management have occurred in recent
years, extending the predicted survival in 2006 to 37 years.2 This
improved prognosis is largely due to the newer pulmonary treatments available
and to the focus on better nutrition.
An organization known as the
CF Foundation (www.cff.org) is ultimately responsible for improving the
quality of life for patients with CF through support of innovative research
and comprehensive care. Maintaining a national patient registry, accrediting
CF care centers, and developing clinical practice guidelines are some ways the
CF Foundation aids in CF management. The CF Foundation's slogan, "Adding
tomorrows every day," provides a clear understanding of the organization's
mission in furthering CF care.
CF is caused by a chromosomal
defect that results in a mutation of the CF transmembrane conductance
regulator (CFTR) gene. CFTR regulates chloride permeability and
sodium transport, leading to obstruction of the exocrine glands by thick,
viscous secretions.3 CF is usually diagnosed in infancy by the
presence of clinical manifestations and an elevated chloride concentration in
induced sweat.4 Patients with CF experience a variety of symptoms,
including persistent cough, frequent lung infections, failure to thrive,
malnutrition, and frequent greasy, bulky stools.
Effective management of CF
requires great commitment on the part of the CF health care team, which
largely includes the patient and family. Recommendations for preventive and
maintenance care for patients with CF have been developed.5
Quarterly outpatient visits consisting of a comprehensive patient history and
physical examination of respiratory, nutritional, and GI manifestations of CF,
along with patient education, are suggested. Recommendations for routine and
annual monitoring of many laboratory values and imaging procedures are also
included in the guidelines. Therapeutic interventions for outpatient (and
inpatient) CF management are discussed in the following sections.
PULMONARY MANAGEMENT
In past years, a
trend in the measurement of lung function (forced expiratory volume in one
second [FEV1]) demonstrated major improvements, with an increase in
the number of patients with mild lung disease and a decrease in prevalence of
severe lung disease.1 Again, this improvement is a result of the
newer CF treatments available for respiratory management.
The main contributors to a
decline in respiratory function in CF include airway obstruction by mucus,
pulmonary inflammation, and infection with various organisms. Cough, sputum
production, chest pain, shortness of breath, and sinus congestion are the
common pulmonary complaints for patients with CF. Possible respiratory
complications include infectious exacerbations, pneumothorax, hemoptysis, and
eventually respiratory failure.
Airway Clearance
Along with drug
therapies and nutrition, airway clearance techniques (ACTs) help maintain and
improve lung function by loosening secretions for expectoration. This is
usually followed by a cough for clearance. Multiple ACTs exist, including
different breathing methods, conventional chest physical therapy with postural
drainage and percussion, and oscillating devices that vibrate the airways
(e.g., Flutter, Acapella) or the entire chest (e.g., Vest). Exercise and
proper hydration may also facilitate ACTs. The sequence of drug therapies
around airway clearance is an important patient education point;
bronchodilators should be taken prior to or with ACTs, and inhaled antibiotics
should be taken after ACTs.
Acute Management
CF pulmonary
exacerbation refers to a change in respiratory signs and symptoms from the
patient's baseline function, necessitating treatment with systemic antibiotics
and augmented airway clearance.5 Generally, adolescents and adults
have more CF exacerbations due to progressive worsening in lung function.1
Common patient complaints associated with pulmonary exacerbation are
increased cough, sputum, shortness of breath, and chest congestion; change in
sputum appearance; decreased appetite; and exercise intolerance.
Infectious Etiologies:
Once a patient with CF has a pulmonary infection caused by an organism, a
cycle may begin between persistent colonization and acute pulmonary
exacerbation. The typical CF pathogens for initial infection are
Staphylococcus aureus and nontypableHaemophilus influenzae,followed
by Pseudomonas aeruginosa (mucoid/nonmucoid), Burkholderia cepacia
, and Stenotrophomonas maltophilia for subsequent infections. S
aureus is the most common cause of respiratory infection in children, and
P aeruginosa is the most common in adolescents and adults.1
Methicillin-resistant S aureus (MRSA) may also be an organism of
concern, particularly for adolescents and adults with CF.1
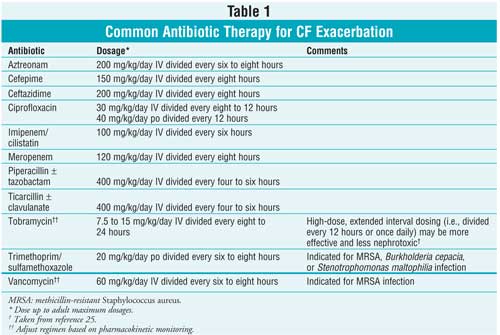
Systemic Antibiotics:
Initial or mild infections may be treated on an outpatient basis with oral
antibiotics; however, subsequent or severe infections usually require
inpatient treatment with intravenous (IV) antibiotics until resolution of
acute symptoms. Antibiotic selection is based on either pathogens typically
isolated in the patient's same age-group or, more preferred, the patient's
previous sputum culture and sensitivity. Generally, until the patient has
cultured P aeruginosa from the sputum, antibiotic therapy effective
against S aureus and H influenzae, such as a
beta-lactamase–stable penicillin or cephalosporin, should be prescribed.
5,6
Once a patient with CF is
infected with P aeruginosa, combination antibiotic therapy is
recommended for synergistic activity and to slow development of resistance.
Typical antibiotic combinations consist of tobramycin plus another
antipseudomonal agent listed in Table 1. However, the patient's regimen
needs to be individualized based on his or her susceptibility patterns.
Management of CF exacerbation in adolescents and adults may be especially
complex given the increase in multidrug-resistant organisms. Duration of
antibiotic treatment is suggested to be at least 10 days, with the usual
course lasting 14 to 21 days.5,6
Home Therapy:
Instead of being hospitalized for a CF exacerbation, some experienced patients
may be candidates for home IV therapy either for the entire duration of
treatment or for part of the course after hospital discharge. Home therapy
requires great family support and understanding, a commitment to ACTs and the
prescribed antibiotic regimen, and use of community resources (e.g., nursing
care) for meeting eligibility for home care of a pulmonary exacerbation.
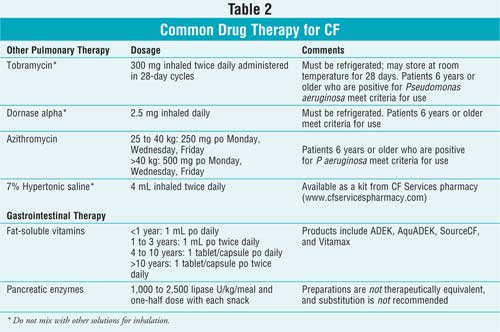
Chronic Management
Chronic, daily
respiratory therapies for CF management may consist of a variety of regimens
from the following drug categories: bronchodilator, mucolytic, inhaled
antibiotic, and anti-inflammatory. The latest studied therapy added to
potential CF drug regimens is hypertonic saline. Table 2 has more
information on the main pulmonary therapies utilized for chronic management of
CF.
Any therapy that is
administered by inhalation, either by nebulization or metered-dose/dry-powder
inhaler is particularly beneficial for patients with CF, compared to other
routes. These formulations usually reach the airway more quickly, more easily,
and in larger drug concentrations.
Bronchodilators:
Short-acting beta-2 agonists (SABAs) such as albuterol and levalbuterol are
frequently prescribed for patients with CF despite the lack of clear research
supporting their use. Potential benefits of SABAs include dilation of the
airways and sputum mobilization. Typical SABA frequencies are scheduled prior
to ACTs two to four times a day.
Historically, theophylline had
a role in CF management. Limited efficacy and concern for toxicity have
reserved its use for patients with documented improvements in lung function.
Mucolytic Therapy:
The abnormal, thick secretions in CF are primarily due to mucus glycoproteins
and DNA. The enzyme DNase I, which digests extracellular DNA, was created in a
human recombinant form known as dornase alpha (Pulmozyme)
inhalation solution. In clinical trials of children and adults with CF,
dornase alpha demonstrated a 28% to 37% reduction in respiratory infections
requiring IV antibiotics, a decrease in hospital stay by 1.4 days, and a mean
increase in FEV1 of 5%. Dornase alpha is recommended for patients 6
years or older; however, it is likely that patients younger than 5 will also
benefit from administration.7
Another nebulized agent
prescribed to decrease mucus viscosity in patients with CF is acetylcysteine.
Its use is limited by inconsistent efficacy data as well as by an
objectionable odor.
Inhaled Antibiotics:
The sputum concentration achieved by aerosol route is much higher than that
by the IV route, which more effectively reduces bacterial density in the lung.
Since its approval in 1998, tobramycin inhalation solution has dramatically
improved lung function in patients with CF. Ramsey and colleagues found a mean
increase in FEV1 of 10% and a reduction of 36% in the need for IV
antibiotics and of 26% for hospitalization in patients using tobramycin
inhalation.8,9
Tobramycin inhalation is
recommended for children 6 years or older colonized with P aeruginosa.
This agent is currently being studied in children as young as 1 year and is
also being studied as a dry-powder inhaler.10,11 Tobramycin for
inhalation has not been researched for the treatment of pulmonary
exacerbations or in patients colonized with B cepacia.
Colistimethate administered
via nebulization is another antibiotic utilized in CF; however, it is
associated with bronchospasm and is not available in an acceptable
formulation. Research evaluating aztreonam lysinate for inhalation and
aerosolized liposomal ciprofloxacin is under way.12,13
Anti-inflammatory
Therapy: Historically, high-dose ibuprofen or oral corticosteroids
have been prescribed for anti-inflammatory management in CF. The use of these
agents is limited by long-term safety concerns. Inhaled corticosteroids may
also be part of the CF drug regimen, but benefit has not been demonstrated in
trials.
Saiman and colleagues
evaluated azithromycin in patients with CF and showed a mean increase of 4.4%
in FEV1, a 47% reduction in days hospitalized, and a 39% reduction
in days on IV antibiotics.14 The exact mechanism of azithromycin in
CF is unclear, although it may be explained by modulation of neutrophil
activity or antagonism of the P aeruginosamucoid biofilm.
Azithromycin is also suggested
for patients 6 years or older colonized with P aeruginosa. Due to
concern for increasing nontuberculous mycobacterial resistance, all patients
initiating azithromycin need appropriate screening. Studies in progress using
azithromycin in CF include patients not infected with P aeruginosa and
patients colonized with B cepacia.15,16
Hypertonic Saline:
Hypertonic saline is thought to improve lung function by increasing airway
hydration, which results in better mucus clearance. A recent Australian trial
was conducted to evaluate this hypothesis in patients older than 6 with CF.
Treatment with hypertonic saline was associated with a reduced frequency of
pulmonary exacerbations and a moderate improvement in lung function.
Currently, there is insufficient evidence to support routine use of hypertonic
saline in CF management, and a larger trial is needed.17
NUTRITIONAL/GI MANAGEMENT
Over 90% of
patients with CF have pancreatic insufficiency requiring enzyme supplement
replacement to achieve adequate nutrition and manage GI symptoms.1
Patients' GI symptoms may include failure to thrive, malnutrition,
gastroesophageal reflux, nausea, flatulence, and steatorrhea from
malabsorption. Meconium ileus, distal intestinal obstruction, and rectal
prolapse are also common GI problems associated with CF.18
To meet the increased caloric
needs in CF, many patients require dietary interventions with oral
supplementation or enteral feeding. Agents used to stimulate appetite in other
diseases (e.g., megestrol acetate, cyproheptadine) are not recommended for
routine use in CF. Growth hormone treatment may be useful in children with CF
to improve growth after aggressive nutritional therapy fails.19
A strong correlation exists
between a higher body mass index and better lung function in children and
adults with CF.1 Therefore, proper nutrition and GI management are
necessary not only for normal growth, but also for comprehensive management of
CF pulmonary disease.
Vitamin Replacement
Fat malabsorption
is common in patients with CF, requiring replacement of the depleted
fat-soluble vitamins. Table 2 provides age-based dosing recommendations
for vitamin A, D, E, and K supplementation.
Other possible minerals and
electrolytes needing replacement in CF include calcium for bone health, iron,
and zinc. Sodium replacement should be suggested during summer months or for
patients living in hot climates.
Pancreatic Enzyme
Replacement
Pancreatic enzyme
supplements were available prior to the Food, Drug, and Cosmetic Act of 1938
and consequently were not completely evaluated for drug efficacy and safety.
This led to two major issues in CF: lack of appropriate dosing information and
substantial variations among different agents. The latter caused worsening of
GI symptoms when enzyme preparations were substituted for one another. This
issue will eventually be resolved because the FDA ruled in 2004 to require
makers of pancreatic enzyme supplements to get their drugs approved within
four years of the rule.21
In the meantime, the CF
Foundation recommends individualized dosing of pancreatic enzymes based on
either ingested fat or weight (see Table 2) titrated to response in
resolution of GI symptoms. Acid suppression with antacids, H2
receptor antagonists, or proton pump inhibitors may increase enzyme efficacy
(and possibly reduce the dosage needed), since the enteric coating requires an
alkaline environment to be dissolved. High dosages (>6,000 lipase U/kg/meal)
of pancreatic enzyme supplements are associated with fibrosing colonopathy and
should be avoided.20
MANAGEMENT OF ASSOCIATED
DISORDERS
Diabetes
The prevalence of
14.3% for CF-related diabetes (CFRD) is likely underestimated due to the lack
of routine diagnostic screening.1 The onset of CFRD occurs during
adolescence; therefore, annual screening beginning at 10 years of age is
recommended. Early disease recognition and treatment is imperative, since poor
glucose control correlates with nutritional failure, worsening pulmonary
disease, and earlier death. The primary cause of CFRD is insulin deficiency,
and as a result, an individualized insulin regimen is most appropriate for
CFRD management.22
Bone Disease
CF-related bone disease occurs in
13.7% of patients, which is also likely an underestimate. Poor nutrition,
vitamin D deficiency, severe lung disease, CFRD, and possible corticosteroid
use are just some of the factors that increase the risk of osteoporosis in
patients with CF. Screening for bone disease should begin at 18 years of age
or as young as 8 with risk factors. Weight-bearing exercise along with calcium
and vitamin D supplementation are recommended for prevention and treatment of
CF-related bone disease. Use of bisphosphonate therapy may be considered in
adults at high risk of fragility fractures.23
Liver and Biliary Tract
Disease
In patients with CF, the prevalence
of liver disease is 9.4%.1 Common conditions in CF are
cholelithiasis that progresses to biliary cirrhosis, hepatic steatosis, and
gallbladder disease. Ursodiol may be beneficial for liver disease by improving
bile flow and displacing toxic bile acids. Chronic therapy with beta-blockers
is useful in CF patients with cirrhosis to prevent variceal hemorrhage.24
Depression
Depression is the
most frequent comorbidity in CF, with a prevalence of 16%. Appropriate
psychosocial interventions in addition to drug therapy may be necessary to
treat depression in adolescents and adults with CF.
CONCLUSION
CF is an inherited
multisystem disease that is characterized by obstruction and infection of the
airways and poor growth. Advances in pulmonary and nutritional management
continue to increase survival in patients with CF. The newer treatments
available along with the agents currently being evaluated will hopefully
continue to "Add tomorrows every day."
References
1. Patient Registry
2005 Annual Report. Bethesda, MD: Cystic Fibrosis Foundation.
2. About Cystic
Fibrosis resource page. Available from: www.cff.org/AboutCF/. Accessed June 9,
2007.
3. Rowe SM, Miller S,
Sorsche EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992-2001.
4. Rosenstein BJ,
Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic
Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132:589-595.
5. Clinical Practice
Guidelines for Cystic Fibrosis. Bethesda, MD: Cystic Fibrosis Foundation.
6. Cystic Fibrosis
Foundation concepts in care consensus conference on microbiology and
infectious disease in cystic fibrosis. Vol V, Section I. May 17-18, 1994.
7. Ramsey BW, Dorkin
HL. Consensus conference: practical applications of Pulmozyme. Pediatr
Pulmonol. 1994;17:404-408.
8. Cystic Fibrosis
Foundation concepts in care consensus conference on use of aerosolized
antibiotics in CF patients. Vol VIII, Section I. September 22-23, 1997.
9. Ramsey BW, Pepe MS,
Quan JM, et al. Intermittent administration of inhaled tobramycin in patients
with cystic fibrosis. N Engl J Med. 1999;340:23-30.
10. Comparison of Two
Treatment Regimens to Reduce PA Infection in Children With Cystic Fibrosis
resource page. Available from: www.clinicaltrials.gov/
ct/show/NCT00097773?order=1. Accessed June 10, 2007.
11. Safety of
Tobramycin Inhalation Powder (TIP) vs Tobramycin Solution for Inhalation in
Patients With Cystic Fibrosis resource page. Available from: www.
clinicaltrials.gov/ct/show/NCT00388505?order=1. Accessed June 10, 2007.
12. Study of Aztreonam
Lysinate for Inhalation (AI) in Cystic Fibrosis Patients With P. Aeruginosa
resource page. Available from: www.clinicaltrials.gov/ct/show/
NCT00128492?order=1. Accessed June 10, 2007.
13. Aradigm receives
FDA orphan drug designation of liposomal ciprofloxacin for cystic fibrosis
[press release]. Available from: phx.corporate-ir.net/phoenix.zhtml?c=
79928&p=irol-newsArticle&ID=853996&highlight. Accessed June 10, 2007.
14. Saiman L, Marshall
BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis
chronically infected with Pseudomonas aeruginosa. JAMA.
2003;290:1749-1756.
15. Effect of
Azithromycin on Lung Function in 6-18 Year-Olds With CF Who Are Not Infected
With Pseudomonas Aeruginosa resource page. Available from: www.
clinicaltrials.gov/ct/show/NCT00431964?order=6. Accessed June 10, 2007.
16. Azithromycin in
Patients With CF, Infected With Burkholderia Cepacia Complex resource
page. Available at:
www.clinicaltrials.gov/ct/show/NCT00298922?order=1. Accessed June 10, 2007.
17. Elkins MR, Robinson
M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline
in patients with cystic fibrosis. N Engl J Med. 2006;354:229-240.
18. Borowitz D, Baker
RD, Stallings V. Consensus report on nutrition for pediatric patients with
cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35:246-259.
19. Hardin DS,
Adama-Huet B, Brown D, et al. Growth hormone treatment improves growth and
clinical status in prepubertal children with cystic fibrosis: results of a
multicenter randomized controlled trial. J Clin Endocrinol Metab.
2006;91:4925-4929.
20. Borowitz D, Grand
RJ, Durie PR. Use of pancreatic enzyme supplements for patients with cystic
fibrosis in the context of fibrosing colonopathy. Consensus Committee. J
Pediatr. 1995;127:681-684.
21. FDA Requires
Pancreatic Extract Manufacturers to Submit Marketing Applications resource
page. Available from: www.fda.gov/bbs/topics/news/2004/NEW01058.html. Accessed
June 11, 2007.
22. Moran A, Hardin D,
Rodman D, et al. Diagnosis, screening, and management of CFRD: a consensus
conference report. J Diabetes Res Clin Pract. 1999;45:61-73.
23. Aris RM, Merkel PA,
Bachrach LK, et al. Consensus conference report: Guide to bone health and
disease in cystic fibrosis. J Clin Endocrinol Metab. 2004; e-published
at: jcem.endojournals.org/cgi/rapidpdf/jc.2004-1629v1.
24. Sokol RJ, Durie PR.
Recommendations for management of liver and biliary tract disease in cystic
fibrosis. Cystic Fibrosis Foundation Hepatobiliary Disease Consensus Group.
J Pediatr Gastroenterol Nutr. 1999;28(Suppl 1):S1-13.
25. Smyth A, Tan KH-V,
Hyman-Taylor P, et al. Once versus three-times daily regimens of tobramycin
treatment for pulmonary exacerbations of cystic fibrosis--the TOPIC study: a
randomized controlled trial. Lancet. 2005;365:573-578.



