US Pharm.
2008;33(12):HS5-HS13.
N
onsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase (COX)-2 inhibitors (COXIBs) are perhaps some of the most extensively used medications in the world.1 In a recent survey of selected prescription and nonprescription drugs recorded during physician office and hospital visits, the U.S. Department of Health and Human Services determined that NSAIDs were the fifth most utilized medication in all age groups.2 The prevalence of at least once-weekly NSAID use in individuals aged 65 and older has been reported to be as high as 70%, with one-half of this group taking at least seven doses per week.3 Finally, preventable NSAID-related hospital admissions have been reported to range from 7% to 11%.4-6 NSAIDs and COXIBs are used for the treatment of fever, inflammation, and pain; in addition to these indications, aspirin also is used for the prevention of vascular events. Unfortunately, NSAIDs and COXIBs are not without adverse effects.
The purpose of this review is to discuss some of the risks associated with NSAID and COXIB use, as well as therapies that can be pursued to prevent or treat morbidities associated with these adverse events. For the purposes of this article, the term NSAIDs, unless otherwise specified, will refer to traditional NSAIDs, aspirin, and COXIBs.
Mechanism of Action of NSAIDs
The inflammatory response occurs when the body is exposed to stimuli such as foreign organisms or antigenic substances.7 Prostaglandins are produced in response to this stimulus. This function can be beneficial in the case of eliminating foreign bacteria, but it can be harmful when it leads to chronic inflammation without resolving the primary detrimental process.7
The principal mechanism of action of the NSAIDs stems from their ability to inhibit the production of prostaglandins.8 Prostaglandins, as well as other mediators such as leukotrienes, are derived from arachidonic acid (AA), which originates from cell-membrane phospholipids through the action of phospholipase A2.8 The metabolism of AA to prostaglandins and leukotrienes is catalyzed by the COX pathway and the 5-lipoxygenase pathway. Cyclooxygenase-1 appears to function as a homeostatic enzyme in most tissues; it allows for maintenance of normal organ function, including normal gastric mucosa, kidney function, and platelet aggregation.9 Cyclooxygenase-2 can be induced by inflammatory stimuli and tends to facilitate the inflammatory response.9 Aspirin and most NSAIDs inhibit both enzymes, whereas COXIBs are highly selective for COX-2.10 Inhibition of COX-1 prevents this normal maintenance from occurring and results in some of the significant side effects caused by aspirin and traditional NSAIDs.8
Gastrointestinal Effects
Gastrointestinal (GI) side effects are common and potentially serious, with as many as 60% of people who use traditional NSAIDs experiencing some type of adverse effect.10-12 Per year, upper-GI complications will develop in 1% to 2% of people using these NSAIDs.13-15 This rate is three to five times higher than in people who do not use these NSAIDs.13-15 The risk of severe complications is even higher in individuals with established risk factors, with a potential case-fatality rate of 5%.16
Damage to the GI tract can occur when the production of prostaglandins is decreased by NSAID inhibition of COX-1. This leads to a decrease in epithelial mucus, the secretion of bicarbonate, mucosal blood flow, epithelial proliferation, and mucosal resistance to injury.17,18 As a result, the normally protective defense mechanisms of the GI mucosa are overwhelmed by such factors as gastric acid, pepsin, and bile salts. NSAIDs also can damage the gastric mucosa through a direct toxicity to the gastric mucosa itself. One of the mechanisms of this injury may be the acidic nature of these medications.8
The GI toxicity associated with nonselective NSAIDs appears to be primarily due to their systemic, not topical, effects.8 As a result, enteric coating on these medications or delivery of the medication in another form (e.g., suppository) would not be expected to decrease their potential for GI toxicity. In addition, use of lower doses of NSAIDs may not decrease GI adverse events. Aspirin doses as low as 30 mg have been shown to decrease prostaglandin synthesis in the gastric mucosa.19 Ibuprofen appears to produce the least risk among older NSAIDs, whereas piroxicam and ketorolac are the worst.1 Newer NSAIDs such as etodolac, meloxicam, and nabumetone are thought to cause less injury to the GI tract.1,20
A variety of risk factors that may increase the likelihood of developing GI side effects secondary to NSAIDs and aspirin have been identified, including prior history of GI event (ulcer, hemorrhage); age greater than 60 years; high dose; concurrent use of corticosteroids; and concurrent use of anticoagulants.20 Other possible risk factors are concomitant use of aspirin and NSAIDs or COXIBs; multiple NSAID use; duration of NSAID use; and Helicobacter pylori status.1,10
Strategies to Reduce GI Toxicity:
Various strategies can be employed to help reduce adverse GI outcomes secondary to traditional NSAIDs (TABLE 1).21-27 First, the use of non-NSAID analgesics should be considered, if feasible. Regular use of acetaminophen has been shown to provide similar analgesia compared with NSAIDs in patients with musculoskeletal conditions.28,29

Misoprostol, a prostaglandin E analog, can help prevent gastric and duodenal ulcers.20,21 Doses necessary to prevent NSAID-induced gastric ulcers have been associated with intolerable abdominal discomfort and diarrhea, however; up to 20% of patients in one study reported diarrhea, and up to a third of patients in another discontinued the medication.22,23
Proton-pump inhibitors (PPIs) are effective for reducing symptomatic and endoscopic ulceration.10 A trial evaluating lansoprazole versus misoprostol for preventing recurrence of gastric ulcers in long-term users of NSAIDs found that efficacy was similar in both groups.22 It was noted that PPIs offer a distinct advantage in that they are dosed once daily and are associated with fewer adverse events.22 Another study compared the efficacy of esomeprazole 20 mg or 40 mg versus placebo for preventing gastric or duodenal ulcers in high-risk patients receiving long-term NSAID treatment, including COX-2–selective NSAIDs.24 For both doses, approximately 95% of patients were ulcer-free at six months.24
Since the primary effect on the gastric mucosa is associated with COX-1, it has been proposed that selective inhibition of COX-2 by COXIBs would provide effective analgesic or anti-inflammatory therapy with minimal effects on the gastric mucosa. Several trials have evaluated the efficacy and toxicity of the COXIBs.30-33 Although they are safer than nonselective NSAIDs, COXIBs are not without GI risk, and this risk may be increased when COXIBs are used concurrently with aspirin. Even when used at a low dose, aspirin has been shown to block COX-1 sufficiently to minimize any GI protection provided by the COXIB.32 Further, COXIBs may be significantly more expensive than traditional NSAIDs plus misoprostol or a PPI. Thus, costs associated with treatment must be assessed before an option is recommended to the patient. Finally, before COXIB therapy is initiated, the patient's cardiovascular (CV) risk must be determined.
CV Effects
NSAIDs can affect the CV system in numerous ways. They can interfere with the antiplatelet activity of aspirin, worsen heart failure (HF), increase blood pressure (BP), and increase the risk of CV disease.34-44
When aspirin binds to COX-1, it acetylates a serine residue that irreversibly inhibits COX-1 for the life of the platelet.34 This decreases the level of COX-1–produced thromboxane A2, a proaggregatory, vasoconstrictive substance. When given prior to aspirin, certain NSAIDs can compete with aspirin for the platelet COX-1 binding site.34,35 The presence of the NSAID prevents the aspirin from binding.
NSAIDs can cause a decrease in serum thromboxane A2 levels, but not irreversibly and only for a portion of the entire dosing interval. Thus, if an NSAID were to be given at the same time as aspirin, this would serve to decrease the complete antiplatelet effect previously invoked by aspirin.34 In a study evaluating the effect of ibuprofen on aspirin's antiplatelet ability, the inhibitory effects of daily low-dose aspirin on platelets were competitively inhibited by the prolonged use of multiple daily doses of ibuprofen (tid dosing), even when aspirin was administered before the first dose of the NSAID.34 Single doses of ibuprofen given two hours before aspirin did not have any sustained effect on platelet activity.34 A similar effect has been demonstrated with naproxen.35 This has yet to be demonstrated with other NSAIDs (such as diclofenac), COXIBs, or acetaminophen. It has been postulated to occur with indomethacin.34
NSAIDs do not cause HF, but they can worsen pre-existing HF.36 NSAID inhibition of prostaglandin synthesis can cause a decrease in renal blood flow and compensatory retention of sodium and water; this increased volume can decrease the effects of diuretics used for HF.36 Systemic vasoconstriction also may occur that can potentially exacerbate the preexisting HF. These are the same mechanisms that can increase BP in patients being treated for hypertension.36 COXIBs have mechanisms similar to those of traditional NSAIDs in the kidney, leading to sodium retention and edema. Therefore, COXIBs would not offer any advantage over traditional NSAIDs in the patient with HF.36
Prostaglandins are converted into a number of prostanoids.37 One of these prostanoids, prostacyclin (produced in the endothelium), causes local smooth-muscle relaxation and vasodilation and also can interact with platelet prostacyclin receptors, thereby antagonizing aggregation.37 COX-2–selective inhibitors prevent the formation of prostacyclin, but they do not prevent the formation of thromboxane A2 (also a proaggregatory, vasoconstrictive product), since only COX-1 enzymes are present on the platelet. It has been postulated that this imbalance of hemostatic compounds may be the reason for the increased risk of CV disease with COXIBs.38,39 Whether nonselective NSAIDs may cause this same imbalance is not yet known.38,39
Four clinical trials, although not principally designed to assess the effects of COX-2 inhibitors on CV outcomes, provide some approximation of risk associated with the use of these agents in various populations.37,40-43 An increased risk of adverse cardiac events was first seen in the Vioxx GI Outcomes Research (VIGOR) study, which compared rofecoxib with naproxen.40 One finding of the study was that rofecoxib was associated with more myocardial infarctions (MIs) than naproxen. The Adenomatous Polyp Prevention on Vioxx (APPROVe) study validated VIGOR's results; compared with placebo, rofecoxib had roughly twice the rate of CV events at 18 months, even though APPROVe investigators tried to exclude patients with increased CV risk.41 A trial of two doses of celecoxib (400 mg/day and 800 mg/day) to prevent colorectal adenomas found that 2.3% of patients receiving 400 mg per day experienced a composite CV endpoint of death from CV causes, nonfatal MI, nonfatal stroke, or nonfatal HF; a 3.4% composite CV endpoint was found in patients taking 800 mg per day.42 Finally, a study of post–coronary artery bypass graft patients comparing valdecoxib or parecoxib with placebo determined that CV events were more common in the COXIB group (2.0%) than in the placebo group (0.5%).43
Traditional NSAIDs, through their ability to increase BP, theoretically could increase the risk of CV events, but the full extent of this potential risk has not been determined. A nested case-control study evaluated the use of celecoxib, rofecoxib, ibuprofen, diclofenac (including combination preparations), naproxen, and other selective (meloxicam, etoricoxib, etodolac, valdecoxib) and nonselective NSAIDs in 9,218 patients to determine the risk of MI.44 An increased risk of MI was evident in patients taking rofecoxib, diclofenac, and ibuprofen despite adjustment for potential confounders, including comorbidities and concurrent use of other drugs. Naproxen was not associated with an increased risk; however, it was not found to be cardioprotective, as was speculated in the VIGOR study.44
In summary, traditional NSAIDs and COXIBs can increase the risk of adverse events in patients who have a history of, or who are at high risk for, CV disease. Evidence is more compelling for the COXIBs, but data do indicate a possible risk with traditional NSAIDs. Until more conclusive data are available, the use of any COX inhibitor or traditional (including OTC) NSAID for a long period of time or at a higher dose should be initiated only in consultation with a physician.37,45
Renal Effects
NSAIDs can bring about two different forms of renal failure. These are hemodynamically mediated failure (due to a reduction in prostaglandin synthesis induced by the NSAID) and acute interstitial nephritis (from a direct toxicity of the drug on the renal parenchyma).46
Renal prostaglandins are vasodilatory in nature.47 Under normal conditions, they appear to exert little influence on renal blood flow and glomerular filtration rate; thus, in healthy patients, NSAIDs probably have minimal effect on renal function.47 However, in volume-depleted or edematous states, these prostaglandins are necessary to compensate for angiotensin- and nor epinephrine-induced renal vasoconstriction.47 When NSAIDs block the production of these prostaglandins in these situations, unopposed vasoconstriction occurs that can lead to acute renal failure.47 Characteristics that may put individuals at increased risk for NSAID-induced renal dysfunction include being elderly; having diabetes, HF, or cirrhosis; dehydration; and blood or fluid loss.46 Specific drugs known to be problematic when combined with anti-inflammatory agents are diuretics, angiotensin-converting enzyme inhibitors, and angiotensin-receptor blockers.
Interstitial nephritis can occur in association with traditional NSAIDs and COXIBs, perhaps due to allergic reaction, direct cellular toxicity, alteration of metabolic pathways, or obstruction.46 Spontaneous recovery usually occurs within weeks to a few months after therapy is discontinued.48
Hepatic Effects
Elevation of liver enzymes can occur with NSAID use, but liver failure is rare.49 Most NSAIDs have been documented to cause liver injury, and the damage tends to be hepatocellular in nature.50,51 The mechanism is not precisely known, but is thought to be immunologically idiosyncratic. Diclofenac and, in particular, sulindac are reported to be more commonly associated with hepatotoxicity. The COXIBs also are linked to hepatotoxicity, although celecoxib is believed to have a lower risk.50 Hepatotoxicity secondary to NSAIDs can occur at any time, but is most likely to happen six to 12 weeks after administration.52 Risk factors for NSAID-induced idiosyncratic hepatotoxicity include female sex, age greater than 50 years, and underlying autoimmune disease.50 Importantly, it is not known whether these are truly risk factors or simply represent the population most likely to use NSAIDs. Another risk factor is concomitant exposure to other hepatotoxic drugs.50
If liver toxicity occurs, the NSAID being used should be discontinued immediately. Once liver function has stabilized, acetaminophen (which has been used in patients who were jaundiced) or aspirin can be used as an alternative.50,53,54 Aspirin is an option because it lacks the diphenylamine ring molecular structure present in NSAIDs that is believed to contribute to their toxicity.55
Hematologic Effects
Hematologic side effects from NSAIDs are related primarily to their antiplatelet activity. As there is no COX-2 enzyme on the platelet, COXIBs would not be expected to produce the same effect.37 Long-term administration of low-dose aspirin is enough to block platelet thromboxane A2 production by more than 95% and to inhibit platelet aggregation ex vivo.56 Conventional nonaspirin NSAIDs inhibit platelet COX reversibly; thus, as stated previously, their effects on platelet aggregation and bleeding time reflect their different half-lives in the circulation.57
Aspirin and other NSAIDs do not cause clinically significant bleeding in most people who use them; this usually occurs in patients with specific conditions (e.g., GI bleed), on certain medications (e.g., anticoagulants), or undergoing elective surgical procedures.57 Nonselective NSAIDs probably should be avoided in patients with underlying qualitative platelet–vessel-wall abnormalities (e.g., von Willebrand's disease or a myeloproliferative disorder), thrombocytopenia, or other inherited coagulation-factor deficiencies (e.g., hemophilia).57
Whether to discontinue aspirin and other NSAIDs preoperatively is controversial, as is whether nonselective NSAIDs cause postoperative bleeding complications.58 Some reports indicate that any significant clinical impact of excessive surgical blood loss after preoperative use of aspirin and other NSAIDs is lessened by cell-salvaging techniques, transfusion, and the use of adjuvant hemostatic agents.57 Recent guidelines suggest that patients on long-term aspirin therapy for coronary or cerebrovascular disease and those who are at risk for coronary events should not discontinue aspirin in the perioperative period unless hemorrhagic complications of the procedure outweigh the risk of an acute thrombotic event.59,60 It is recommended that aspirin be continued in patients at high risk for cardiac events who are undergoing noncardiac surgery or percutaneous coronary intervention.60 Aspirin can be continued in patients undergoing minor dental procedures, minor dermatologic procedures, or cataract surgery.60 It is recommended that aspirin be continued in patients requiring surgery within six weeks of having a bare metal coronary stent placed or within 12 months of having a drug-eluting stent placed.59 In patients not at high risk for cardiac events who are undergoing a noncardiac procedure, it is recommended that aspirin and aspirin-containing medications be stopped seven to 10 days before surgery.60 Nonaspirin, nonselective NSAIDs (and probably COXIBs) should be discontinued five half-lives before surgery.60 A list of these medications appears in TABLE 2.60

Neutropenia is another, albeit rare, complication of NSAID therapy. A case-control study indicated an increased risk of neutropenia with NSAID use; however, no particular risk factors were determined, and no specific NSAID was identified as being the main cause.61
Effects on Pregnancy
The use of NSAIDs around the time of conception may be associated with a risk of miscarriage.62 The proposed mechanism is that, by blocking prostaglandin production, NSAIDs can interfere with prostaglandins' role in implantation, thus possibly leading to abnormal implantation and miscarriage.62 For women who are trying to conceive, it would be prudent to avoid NSAID use and to consider alternative analgesics, such as acetaminophen, instead. Finally, it is important that these agents be discontinued during the third trimester of pregnancy. This helps prevent problems such as prolonged gestation and labor, increased bleeding, and premature closure of the ductus arteriosus.63
Role of the Pharmacist
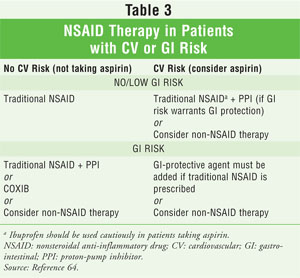
One of the most important areas a pharmacist can act on with regard to the safe and effective use of NSAIDs is risk management. This is perhaps most evident in patients who have GI or CV risk factors (TABLE 3).64 Once the pharmacist has determined that NSAID therapy is appropriate, he or she should counsel the patient to facilitate the prevention or mitigation of other adverse events. Finally, NSAIDs can interact with many medications. It is important for pharmacists to know their patients well, be familiar with which medications they are taking, and understand how these medications can potentially interact with NSAIDs.
REFERENCES
1. Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology. 2001;120:594-606.
2. National Center for Health Statistics. Health, United States, 2007 with chartbook on trends in the health of Americans. www.cdc.gov/nchs/data/hus/hus07.pdf. Accessed August 12, 2008.
3. Talley NJ, Evans JM, Fleming KC, et al. Nonsteroidal anti-inflammatory drugs and dyspepsia in the elderly. Dig Dis Sci. 1995;40:1345-1350.
4. van der Hooft CS, Dieleman JP, Siemes C, et al. Adverse drug reaction-related hospitalisations: a population-based cohort study. Pharmacoepidemiol Drug Saf. 2008;17:365-371.
5. Howard RL, Avery AJ, Slavenburg S, et al. Which drugs cause preventable admissions to hospital? A systematic review. Br J Clin Pharmacol. 2006;63:136-147.
6. Kongkaew C, Noyce PR, Ashcroft DM. Hospital admissions associated with adverse drug reactions: a systematic review of prospective observational studies. Ann Pharmacother. 2008;42:1017-1025.
7. Furst DE, Ulrich RW. Nonsteroidal anti-inflammatory, disease-modifying antirheumatic drugs, nonopioid analgesics and drugs used in gout. In: Katzung BG, ed. Basic and Clinical Pharmacology. 10th ed. New York, NY: McGraw-Hill Medical; 2007.
8. Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888-1899.
9. Needleman P, Isakson PC. The discovery and function of COX-2. J Rheumatol. 1997;24(suppl 49):6-8.
10. Jones R, Rubin G, Berenbaum F, Scheiman J. Gastrointestinal and cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Med. 2008;121:464-474.
11. Singh G, Ramey DR, Morfeld D, et al. Gastrointestinal tract complications of nonsteroidal anti-inflammatory drug treatment in rheumatoid arthritis. A prospective observational cohort study. Arch Intern Med. 1996;156:1530-1536.
12. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241-249.
13. García Rodríguez LA, Jick H. Risk of upper gastrointestinal bleeding and perforation associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343:769-772.
14. Langman MJ, Weil J, Wainwright P, et al. Risks of bleeding peptic ulcer associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343:1075-1078.
15. Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med. 1991;115:787-796.
16. Targownik LE, Nabalamba A. Trends in management and outcomes of acute nonvariceal upper gastrointestinal bleeding: 1993-2003. Clin Gastroenterol Hepatol. 2006;4:1459-1466.
17. Whittle BJR. Mechanisms underlying gastric mucosal damage induced by indomethacin and bile-salts, and the actions of prostaglandins. Br J Pharmacol. 1977;60:455-460.
18. Wolfe MM, Soll AH. The physiology of gastric acid secretion. N Engl J Med. 1988;319:1707-1715.
19. Lee M, Cryer B, Feldman M. Dose effects of aspirin on gastric prostaglandins and stomach mucosal injury. Ann Intern Med. 1994;120:184-189.
20. Lanza FL. A guideline for the treatment and prevention of NSAID-induced ulcers. Members of the Ad Hoc Committee on Practice Parameters of the American College of Gastroenterology. Am J Gastroenterol. 1998;93:2037-2046.
21. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroid al anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241-249.
22. Graham DY, Agrawal NM, Campbell DR, et al. Ulcer prevention in long-term users of nonsteroidal anti-inflammatory drugs: results of a double-blind, randomized, multicenter, active- and-placebo-controlled study of misoprostol vs lansoprazole. Arch Intern Med. 2002;162:169-175.
23. Fendrick AM, Bernstein SJ, Scheiman JM. Prevention of NSAID induced ulcers in the elderly. Geriatrics. 2005;Oct (suppl):S1-S14.
24. Scheiman JM, Vakil N, Hawkey CJ, et al. Esomeprazole prevents gastric and duodenal ulcers in at-risk patients on continuous non-selective or COX-2-selective NSAID therapy. Gastroenterology. 2004;126(suppl 2):A-82 [abstract].
25. Regula J, Butruk E, Dekkers CP, et al: Prevention of NSAID-associated gastrointestinal lesions: a comparison study pantoprazole versus omeprazole. Am J Gastroenterol. 2006;101:1747-1755.
26. Hawkey CJ, Karrasch JA, Szczepañski L, et al. Omeprazole compared with misoprostol for ulcers associated with nonsteroidal antiinflammatory drugs. N Engl J Med. 1998;338:727-734.
27. Irani S, Krevsky B, Desipio J, et al. Rapid protection of the gastroduodenal mucosa against aspirin-induced damage by rabeprazole. Aliment Pharmacol Ther. 2008;27:498-503.
28. Woo WW, Man SY, Lam PK, Rainer TH. Randomized double-blind trial comparing oral paracetamol and oral nonsteroidal antiinflammatory drugs for treating pain after musculoskeletal injury. Ann Emerg Med. 2005;46:352-361.
29. Superio-Cabuslay E, Ward MM, Lorig KR. Patient education interventions in osteoarthritis and rheumatoid arthritis: a meta-analytic comparison with nonsteroidal antiinflammatory drug treatment. Arthritis Care Res. 1996;9:292-301.
30. Simon LS, Weaver AL, Graham DY, et al. Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis: a randomized controlled trial. JAMA. 1999;282:1921-1928.
31. Emery P, Zeidler H, Kvien TK, et al. Celecoxib versus diclofenac in long-term management of rheumatoid arthritis: randomised double-blind comparison. Lancet. 1999;354:2106-2111.
32. Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. JAMA. 2000;284:1247-1255.
33. Bensen WG, Fiechtner JJ, McMillen JI, et al. Treatment of osteoarthritis with celecoxib, a cyclooxygenase-2 inhibitor: a randomized controlled trial. Mayo Clin Proc. 1999;74:1095-1105.
34. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
35. Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295-1301.
36. Amabile CM, Spencer AP. Keeping your patient with heart failure safe: a review of potentially dangerous medications. Arch Intern Med. 2004;164:709-720.
37. Bennett JS, Daugherty A, Herrington D, et al. The use of nonsteroidal anti-inflammatory drugs (NSAIDs): a science advisory from the American Heart Association. Circulation. 2005;111:1713-1716.
38. FitzGerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709-1711.
39. Topol EJ. Failing the public health--rofecoxib, Merck, and the FDA. N Engl J Med. 2004;351:1707-1709.
40. Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343:1520-1528.
41. Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-1102.
42. Solomon SD, McMurray JJV, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071-1080.
43. Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081-1091.
44. Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ. 2005;330:1366-1372.
45. FDA. Public health advisory. Non-steroidal anti-inflammatory drug products (NSAIDS). www.fda.gov/cder/drug/advisory/nsaids.htm. Accessed August 7, 2008.
46. House AA , Silva Oliveira S, Ronco C. Anti-inflammatory drugs and the kidney. Int J Artif Organs. 2007;30:1042-1046.
47. Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357-377.
48. Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal antiinflammatory drugs. N Engl J Med. 1984;310:563-572.
49. Abraham PA, Keane WF. Glomerular and interstitial disease induced by nonsteroidal anti-inflammatory drugs. Am J Nephrol. 1984;4:1-6.
50. O'Connor N, Dargan PI, Jones AL. Hepatocellular damage from non-steroidal anti-inflammatory drugs. Q J Med.2003;96:787-791.
51. Aithal GP, Day CP. Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin Liver Dis. 2007;11:563-575.
52. Aithal PG, Day CP. The natural history of histologically proved drug induced liver disease. Gut. 1999;44:731-735.
53. Prescott LF. Effect of non-narcotic analgesics on the liver. Drugs. 1986;32(suppl 4):129-147.
54. Jones AL. Recent advances in the management of late paracetamol poisoning. Emerg Med. 2000;12:14-21.
55. Boelsterli UA, Zimmerman HJ, Kretz-Rommel A. Idiosyncratic liver toxicity of nonsteroidal anti-inflammatory drugs: molecular mechanisms and pathology. Crit Rev Toxicol. 1995;25:207-235.
56. FitzGerald GA, Oates JA, Hawiger J, et al. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest. 1983;71:676-688.
57. Schafer AI. Effects of nonsteroidal anti-inflammatory therapy on platelets. Am J Med. 1999;106:25S-36S.
58. Russell MW, Jobes D. What should we do with aspirin, NSAIDs, and glycoprotein-receptor inhibitors? Int Anesthesiol Clin. 2002;40:63-76.
59. Backman SB, Bondy RM, Deschamps A, et al. Perioperative considerations for anesthesia. In: Souba WW, Fink MP, Jurkovich GJ, et al, eds. ACS Surgery: Principles & Practice. 6th ed. New York, NY: WebMD Professional Publishing; 2008.
60. Douketis JD, Berger PB, Dunn AS, et al. The perioperative management of antithrombotic therapy: American College of Chest Physicians evidence-based clinical practice guidelines (8th ed). Chest. 2008;133(suppl 6):299S-339S.
61. Strom BL, Carson JL, Schinnar R, et al. Nonsteroidal anti-inflammatory drugs and neutropenia. Arch Intern Med.1993;153:2119-2124.
62. Li DK, Liu L, Odouli R. Exposure to non-steroidal anti-inflammatory drugs during pregnancy and risk of miscarriage: population based cohort study. BMJ. 2003;327:368-372.
63. Janssen NM, Genta MS. The effects of immunosuppressive and anti-inflammatory medications on fertility, pregnancy, and lactation. Arch Intern Med. 2000;160:610-619.
64. Fendrick AM. COX-2 inhibitor use after Vioxx: careful balance or end of the rope? Am J Manag Care. 2004;10:740-741.


