US Pharm. 2007;32(6):45-53.
Alzheimer's disease (AD) is the most common form of dementia in the elderly, accounting for 50% to 75% of patients with dementia.1-3 Dementia is characterized by memory impairment and at least one of the following: aphasia, apraxia, agnosia, or a disturbance in the ability to think abstractly and to plan, initiate, sequence, monitor, and stop complex behavior.2 More than four million people have AD in the United States.3-6 The prevalence of dementia increases dramatically with age, affecting about 5% to 8% of individuals older than 65, 15% to 20% of individuals older than 75, and 25% to 50% of individuals older than 85.2 Given projections for the continued growth in the elderly population, the prevalence of AD will likely quadruple over the next 50 years unless effective means of prevention can be found.7
Beyond age, confirmed risk factors for AD include dementia in a close family member and presence of the apolipoprotein (APO) E4 allele.8 APO E, which is controlled by chromosome 19, is synthesized and secreted by astrocytes and internalized by neurons, ultimately affecting the phosphorylation of tau protein.9 The three major types of
Pathophysiology
The exact pathophysiology of AD remains unknown; however, the classic neuropathologic features of AD include deposition of extracellular beta-amyloid plaques, formation of intracellular neurofibrillary tangles, and neuronal degeneration.1,10 Accumulation of beta-amyloid in the brain is considered the primary influence driving the pathogenesis of AD.11 Extracellular deposition of insoluble beta-amyloid protein contributes to neuritic plaque formation and may be associated with inflammation of the surrounding tissue, oxidative stress, synaptic dysfunction, deficits in neurotransmission, and the formation of neurofibrillary tangles.1,10,12
Neurofibrillary tangles contain paired helical filaments that aggregate in dense bundles. These paired helical filaments are formed from tau protein, which provides the structural support to microtubules, which in turn serve as the skeletal support system for neurons. When tau protein is hyperphosphorylated, it is unable to bind effectively to microtubules, and the microtubules collapse, ultimately leading to cell death and the formation of neurofibrillary tangles.1,10,12 The number of neurofibrillary tangles is correlated with the severity of dementia.
Accumulation of beta-amyloid plaques and the subsequent formation of neurofibrillary tangles are accompanied by neuronal loss and atrophy in brain areas important for cognition. Such neuronal damage can ultimately lead to cognitive impairment.1,10,12
Diagnosis
A diagnosis of probable AD should be made only when other etiologies for dementia have been ruled out. A definitive diagnosis of AD depends on microscopic examination of the brain at autopsy, which reveals numerous beta-amyloid plaques outside neurons and neurofibrillary tangles within neurons widely distributed in the cerebral cortex. A clinical diagnosis using the National Institutes of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria and/or the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition criteria for AD conforms to the pathologic diagnosis 70% to 90% of the time.2
Advantages of neuroimaging over neuropsychological testing include increased diagnostic accuracy, freedom from cultural bias for interpretation, independence from level of education, and rater-independent objective measures of brain anatomy and function. The major disadvantage of neuroimaging is cost.13 Cross-sectional imaging techniques, such as computed tomography (CT) and magnetic resonance imaging (MRI), are noninvasive methods for detecting cortical atrophy. Data from CT and MRI provide accuracy in the diagnosis of AD 88% and 87% of the time, respectively.9 Studies using volumetric MRI to measure the size of key brain structures have suggested that a smaller size of these structures in patients with mild cognitive impairment can identify patients who will progress to AD within three years.14 Positron emission tomography (PET), using the FDG tracer, provides accuracy in the diagnosis of AD 89% of the time. A new PET tracer, 18F-FDDNP, crosses the blood-brain barrier and binds to beta-amyloid plaques and neurofibrillary tangles, allowing for the identification of neuropathologic findings in living patients with AD. Single-photon emission CT and magnetic resonance spectroscopy provide accuracy in the diagnosis of AD 72% and 91% of the time, respectively. These imaging techniques will likely be used more frequently in the future to improve the accuracy of diagnosing AD, to identify patients at risk for developing AD, and to monitor the effect of therapeutic agents currently under development to modify AD progression.9
Progression of AD
The progression of AD is gradual, with an average duration between the onset of symptoms and death of eight to 10 years.2 In clinical practice, the Mini-Mental State Examination (MMSE) is the most practical tool to establish baseline cognitive function and evaluate changes in severity of AD. MMSE scores range from 0 to 30 points, with higher scores indicating less impairment. A score between 10 and 26 is typical of mild-to-moderate AD. The average expected decline in MMSE score in an untreated patient is 2 to 4 points per year, so a decline in MMSE score of less than 2 to 4 points per year may be used as an indicator of treatment success.15
Pharmacotherapy for Cognitive Symptoms
The treatment of AD is guided by the stage of illness and the specific symptoms manifested by the patient. Treatment goals for the management of AD-related cognitive symptoms include improved quality of life, mood, and behavior; maximization of function; minimization of further decline of cognitive and functional losses associated with dementia; and improved comfort of patients and families in the context of living with a difficult disease.2
Acetylcholinesterase Inhibitors: Acetylcholine and nicotinic receptors play an important role in both memory and learning.3,10 The body contains two cholinesterases, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). As AD progresses, AChE activity decreases and BuChE activity increases. At advanced stages of AD, BuChE may replace AChE in hydrolyzing acetylcholine in the brain.3 Cholinergic synapses are lost early in AD, a finding that has led to the development of AChE inhibitors (AChEIs). AChEIs prevent the breakdown of acetylcholine by AChE and improve cholinergic neurotransmission in surviving neurons.10
AChEIs were the first pharmacologic treatments for AD to be approved by the FDA. Four AChEIs--donepezil (Aricept), galantamine (Razadyne), rivastigmine (Exelon), and tacrine (Cognex)--are approved for the treatment of mild-to-moderate AD. The use of tacrine is limited due to risks of hepatotoxicity and the inconvenience of a four times daily dosing schedule.3 Donepezil is also available in an orally disintegrating tablet, which is useful for patients who have difficulty swallowing tablets.16,17
Donepezil and galantamine selectively inhibit AChE, whereas rivastigmine inhibits both AChE and BuChE. All AChEIs interact with nicotinic receptors and/or muscarinic receptors via acetylcholine, which is made more available by cholinesterase inhibition. Galantamine also binds to a site on the nicotinic receptor, which is different from the binding site of acetylcholine, and amplifies the actions of acetylcholine at postsynaptic and presynaptic nicotinic receptors. Further investigation is required to more clearly describe the clinical consequences of the differences in pharmacologic properties among the AChEIs.3
Despite differences in pharmacodynamic properties and mechanisms of action, individual AChEIs appear to have a similar magnitude of benefit. Therefore, donepezil, galantamine, and rivastigmine are all considered first-line agents.3,14 Overall, clinical trials using AChEIs have demonstrated a modest yet significantly favorable effect on global and cognitive functioning, activities of daily living, behavioral symptoms, caregiver burden, and resource utilization in patients with mild-to-moderate AD.3,18,19 Although patients with AD, on average, decline to their pretreatment level after nine to 12 months of therapy, they function significantly better than the placebo-treated group at that point. Since AChEIs exhibit a dose response, patients should be titrated to the maximum dosage of an AChEI with respect to adverse effects (Table 1).18 The most common side effects of AChEIs are nausea, vomiting, diarrhea, abdominal pain, dizziness, and anorexia.3,6,16 These side effects occur more frequently during the titration of dosages. Gastrointestinal side effects can be minimized with gradual dose increases and administration with food. If intolerable side effects occur with an AChEI, treatment should be discontinued for at least one week or until adverse effects resolve. An alternative AChEI may then be prescribed.3,16
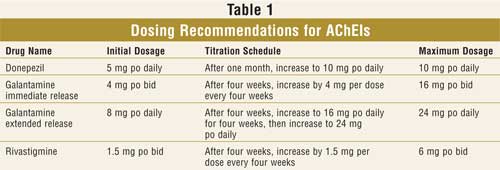
Treatment with an AChEI should be initiated once a diagnosis of probable AD is established. Patients who show improvement or stabilization during the initial three to six months of treatment are considered to have experienced benefit from treatment. Open-label follow-up studies show that the effects of AChEIs continue for at least two years, although some may experience a response to treatment beyond two years. Therefore, once benefit has been established in a patient with AD, the AChEI should be continued for at least one to two years unless intolerable adverse effects occur or until the patient or caregiver requests that treatment be discontinued.18
Limited data show significant benefits of AChEIs in patients with more severe AD. There is no indication that these effects are less for patients with lower MMSE scores at the beginning of treatment. The effects of AChEIs do not stop simply because the patient progresses to a severe stage of AD.3,18
The decision to discontinue AChEI therapy in patients with AD should be made with consideration to both efficacy and severity of side effects. Patients who show improvement or stabilization during the initial three to six months of treatment are considered to have benefited from treatment. Deciding whether to continue treatment in patients who continue to deteriorate is more difficult. Since AD is a progressive disease, patients may experience less disease progression with treatment than they would without treatment. If there are doubts about the clinical benefit of therapy after one to two years, the AChEI may be discontinued for four weeks. If the patient worsens within a few weeks of discontinuing therapy, the patient should resume treatment; however, if the patient does not experience a change in clinical condition during the four weeks, the AChEI may be permanently discontinued. Switching to another therapy may be considered at this point.3,11,18
N-methyl-d-aspartate Receptor Antagonists: N-methyl-d-aspartate (NMDA) receptor–mediated glutamatergic neurotransmission has been shown to be necessary in animal models of learning and memory. Chronic over-stimulation of glutamatergic NMDA receptors results in excess calcium, causing osmotic disturbances and inappropriate activation of catabolic enzyme pathways and, ultimately, leading to neuronal death. Such cell death may trigger a chain reaction of neuronal loss due to release of additional glutamate from dying cells. Therefore, excitotoxicity may contribute to the widespread loss of neurons in AD.3,10 On the other hand, physiologic activation of the NMDA receptor appears to be necessary for normal cognitive function. Therefore, medications that finely modulate NMDA receptor activity have been shown to be effective in the treatment of AD.3
Memantine (Namenda), a low-affinity NMDA receptor antagonist, is approved for the treatment of moderate-to-severe AD. It appears to inhibit the pathologic neural toxicity associated with prolonged glutamate release without blocking physiologic activation of the NMDA receptor.3,10 Memantine has been shown to delay cognitive and functional decline in patients with moderate-to-severe AD, with a side effect profile similar to that of placebo. In addition, studies suggest that memantine plus donepezil is significantly more efficacious than donepezil alone.3,20 Therefore, treatment with memantine as an adjunct therapy to AChEIs is reasonable in patients with moderate-to-severe AD.6 The recommended oral dosage titration is 5 mg daily for one week, then 5 mg twice daily for one week, then 10 mg in the morning and 5 mg in the evening for one week, and then 10 mg twice daily.
Vitamin E:According to the oxidative stress hypothesis, AD may result from acceleration of the aging process in specific brain regions that are exposed to free radicals. Free radicals cause lipid peroxidation in cells that alter and cleave fatty acid side chains. Lipid peroxidation may lead to cell death by destroying membrane integrity, rupturing organelles, or rupturing the cell. The antioxidant protective system becomes less efficient with age, and cell membranes become more vulnerable to oxidative damage.21
Vitamin E is a fat-soluble vitamin, which acts as an antioxidant and an anti-inflammatory agent. Studies have shown decreased vitamin E levels in the blood and cerebrospinal fluid of patients with AD.21 One study demonstrated that patients with moderate to severe AD who were treated with vitamin E 1,000 IU twice daily experienced significant delays in reaching the combined end point of death, institutionalization, loss of ability to perform basic activities of daily living, and rating of severe dementia.22
The American Psychiatric Association "Practice Guidelines for Treatment of Patients with Alzheimer's Disease and Other Dementias of Later Life" states that vitamin E may be considered for patients with moderate AD to slow the rate of disease and might also be beneficial earlier or later in the course of the disease.2 Recently, a meta-analysis found that vitamin E dosages of 400 IU daily or greater may increase all-cause mortality.16 Therefore, the decision to use vitamin E in patients with AD should include the patient and caregiver.
Selegiline: Selegiline (Eldepryl) is a selective monoamine oxidase type B inhibitor that is approved for the treatment of Parkinson's disease. Selegiline decreases the degradation of catecholamines in the brain and thus reduces the production of oxygen-free radicals, which may lead to neuroprotective effects.11 A randomized, placebo-controlled trial comparing the effect of vitamin E, selegiline, selegiline plus vitamin E, and placebo in patients with AD over a two-year period demonstrated a significant delay in time to death, placement in a nursing home, development of severe dementia, and/or impairment of activities of daily living in the patients treated with vitamin E and/or selegiline compared with those treated with placebo. Selegiline is associated with orthostatic hypotension, anxiety, irritability, and a risk for significant drug-drug interactions; therefore, vitamin E, which appeared to be equally efficacious, is preferable. Because no evidence of an additive effect of vitamin E and selegiline was found, the authors determined that there is no empirical basis for using the two agents in combination.2,17
Ginkgo Biloba: The extract of the Ginkgo biloba tree, EGb 761, is a popular substance used in
A 52-week, randomized, double-blind, placebo-controlled, multicenter study conducted by the North American EGb Study Group compared EGb 120 mg orally once daily to placebo in 309 patients with mild-to-moderate AD. Results of this study showed no significant differences in the reporting of adverse events with EGb compared with placebo. Patients treated with EGb experienced significantly less decline in cognitive and social functioning, compared with those treated with placebo.23 A 24-week randomized, double-blind, placebo-controlled, multicenter study conducted in Germany compared EGb 120 mg orally twice daily to placebo in 222 patients with mild-to-moderate AD or multi-infarct dementia. Patients treated with EGb experienced significant improvement in cognitive functioning compared to patients treated with placebo. There was no significant difference in activities of daily living between the groups.24 Ginkgo biloba extract may increase the risk of bleeding in patients taking aspirin, NSAIDs, or warfarin. Side effects of Ginkgo biloba include headache, dizziness, heart palpitations, allergic dermatitis, and gastrointestinal adverse effects.
Estrogen: Another strategy for the prevention and treatment of AD is estrogen. Estrogen reduces both the production and deposition of beta-amyloid. Clinical trials reveal that the age at which hormone replacement therapy (HRT) is initiated affects the incidence of AD in women.25
Zandi et al. showed that women who started estrogen therapy at the time of menopause and continued therapy for 10 years experienced a lower incidence of AD, whereas women who began estrogen therapy at age 65 or older demonstrated an increased incidence of AD. The Women's Health Initiative Memory Study showed an increased incidence of AD in women started on conjugated estrogens/methylprogesterone at age 65 or older. Collectively, the data from these trials show that women who began HRT or estrogen therapy 10 years or more after menopause had an increased risk of developing AD. However, women who began HRT at the time of transition into menopause showed a reduced risk of developing AD.25
While existing data on the role of estrogen therapy in preventing AD are encouraging, data on its role in treating women with existing disease are not. Data from large, randomized, double-blind, placebo-controlled, parallel-group trials show that both short- and long-term estrogen therapy does not improve ADsymptoms in most women, despite the positive benefits reported in smaller open-label clinical trials. Estrogen appears to maintain and sustain neuronal viability to prevent degenerative disease but is ineffective in reversing the degenerative disease process.25
Currently, estrogen therapy is not recommended for the treatment or prevention of AD.17 The challenge remains to develop a therapeutic strategy for promoting the beneficial effects of estrogen in the brain while preventing consequences of unopposed estrogen in other organ systems, such as increased risk of uterine cancer.25
HMG-CoA Reductase Inhibitors: Medications that inhibit HMG-CoA reductase have been proven to reduce serum cholesterol and LDL cholesterol. Lovastatin (Mevacor) and simvastatin (Zocor) are able to cross the blood-brain barrier into the central nervous system (CNS), while others, such as atorvastatin (Lipitor), fluvastatin (Lescol), pravastatin (Pravachol), and rosuvastatin (Crestor), are not able to do so. HMG-CoA reductase inhibitors are thought to reduce levels of certain peptides that can lead to the formation of amyloid plaques. By reducing the amount of these peptides, HMG-CoA reductase inhibitors may reduce the incidence or the progression of AD; however, there is currently insufficient evidence to show that HMG-CoA reductase inhibitors reduce the progression of AD.4
NSAIDs: NSAIDs may protect against AD by decreasing inflammatory processes in the brain, reducing cell death, and/or decreasing the formation of beta-amyloid plaques. Results of a meta-analysis reveal a reduction in risk of 26% for patients who have used NSAIDs at some point during their lifetime and a reduction in risk of 58% for patients who have used NSAIDS for two or more years. These findings suggest that NSAIDs are more likely to reduce the risk of AD when exposure is sustained.7
The benefit of NSAIDs in AD remains controversial. Currently, the AD Anti-Inflammatory Prevention Trial is evaluating the use of naproxen, celecoxib, or placebo in cognitively normal elderly patients over a seven-year period, with the primary outcome being a diagnosis of AD.1,7
New Horizons for Treatment of AD
Most likely, beta-amyloid plaques precede the onset of clinical signs by a long preclinical time period. Beta-amyloid accumulation precedes the formation of neurofibrillary tangles, neurodegeneration, and functional loss. Therefore, the removal of beta-amyloid or the prevention of beta-amyloid accumulation may not only correct beta-amyloid–related toxicity but also prevent the formation of neurofibrillary tangles.12 Currently, there are no approved treatments that will stop the functional deterioration of AD. Successful disease-modifying therapies are needed to address the pathogenesis of AD. Approaches to modifying beta-amyloid accumulation include inhibiting its production, blocking its aggregation and toxicity, and stimulating its degradation and removal.26
Beta-amyloid is cleaved from the beta-amyloid precursor protein by the enzymes beta- and gamma-secretase. Therefore, inhibition of these enzymes would decrease the production of beta-amyloid. Selective inhibitors of gamma-secretase result in the accumulation of a potentially neurotoxic substance; therefore, researchers are trying to identify a potent beta-secretase inhibitor.26
Another pharmacologic strategy is to prevent the aggregation and toxicity of beta-amyloid. Developing a compound that binds to beta-amyloid and shields it may prevent aggregation of beta-amyloid. Selectively blocking beta-amyloid ion conduction might prevent the neurotoxicity of beta-amyoid.26
Active immunization with beta-amyloid causes the formation of antibodies against beta-amyloid plaques in patients with AD. Mechanisms used to describe the therapeutic effect of immunotherapy include microglia-mediated removal of beta-amyloid, removal by the peripheral beta-amyloid sink, and antibody-mediated disaggregation of beta-amyloid. Microglia act as the immune cells of the CNS and appear to be highly mobile. They have an important role in the removal of beta-amyloid. The peripheral beta-amyloid sink hypothesis refers to antibodies binding to beta-amyloid in the blood outside the CNS. The blood then serves as a sink for beta-amyloid, pulling it from the brain and shifting the equilibrium in favor of the soluble protein over the plaque-associated, insoluble form. Antibodies against beta-amyloid may reduce beta-amyloid related pathology, neurofibrillary tangles, and neurodegeneration.12
The first active vaccine, AN-1792, consisted of beta-amyloid fibrils generated by aggregation of synthetic beta-amyloid peptides with an adjuvant. In a phase II clinical trial, AN-1792 was given to 298 patients with AD. The immunization schedule involved injections into the deltoid muscle of 225 mcg AN-1792 with 50 mcg QS-21 (an immunogenic adjuvant) at baseline and after one, three, six, nine, and 12 months.27 This trial was suspended only a few months into the study due to episodes of meningoencephalitis in 18 of the vaccinated patients.5,12 Although no further inoculations were administered, patients who were immunized were followed for two years after the initial vaccination. Both cognitive function and activities of daily living declined less in patients with increased serum titers of antibodies against beta-amyloid. In addition, a correlation was observed between level of antibody titers and clinical outcomes. Those with the highest levels of antibodies against beta-amyloid remained cognitively and functionally stable for the entire two-year period; antibody titers also remained increased.12,28 The cases of meningoencephalitis raised the hypothesis that clinical inflammation was required to clear beta-amyloid. However, data from a neuropathologic analysis of four brains obtained at autopsy did not support this speculation. Results from the autopsies showed reduced beta-amyloid pathology, reduced astrogliosis, and maintained tangle pathology. Moreover, removal of beta-amyloid was observed in the absence of prior episodes of meningoencephalitis, suggesting that meningoencephalitis is not required for beta-amyloid removal. In addition to improving neuropathology, antibodies against beta-amyloid are effective in slowing the clinical progression of AD.12
Advantages of active immunization include lower costs and less frequent administration. Concerns regarding active immunization for AD include the fact that vaccination against ubiquitous and naturally occurring beta-amyloid might cause a widespread inflammatory response. This inflammatory response may resemble what is seen in autoimmune disease.27 Once an active immunization is administered, there is potential for adverse reactions associated with runaway immune responses. An advantage of passive immunization is the control of the dosage of antibody in circulation by adjusting the dosage and frequency of administration. Passive immunization can also overcome inadequate immune responses to active immunizations. Passive vaccines to beta-amyloid, as well as active vaccines that are based on shortened beta-amyloid peptide, have been successfully tested in mice and have already been proclaimed as a safer alternative for treating AD than AN-1792.5,27,28
Since beta-amyloid can be imaged by PET in living people, detection of beta-amyloid could identify patients at high risk for developing clinical signs of AD. If clinical safety can be improved, immunotherapy against beta-amyloid could be a future option for treatment of AD as well as for prevention of AD in high-risk patients.5,12
Role of the Pharmacist
Caregiver burden results in poor health as evidenced by a greater number of illnesses and psychological distress. Caregivers often feel overwhelmed by the hard work and personal loss associated with caring for a patient with AD. Pharmacists can help by encouraging caregivers to participate in support groups for caregivers of patients with AD.17 In addition to providing helpful information about AD as well as information about caring for patients with AD and methods for decreasing caregiver burden, these groups may enhance the quality of life of caregivers and delay nursing home placement. Support groups may vary in their approaches; therefore, caregivers may try several before finding one that fits their needs. The Alzheimer's Association is also a helpful resource for caregivers.2
As AD progresses, patients often lose the ability to make medical, legal, and financial decisions. Documents such as durable power of attorney for health care and financial matters can help families avoid the difficulty and expense of petitioning for guardianship. Patients should be advised to discuss preferences about medical treatment with their families early in the course of the disease while the patient is capable of making his or her desires known. Patients with more complex financial issues should be referred to an attorney or financial planner.2
When families feel they are no longer able to provide care at home, they may need to place the patient in a long-term care facility. This possibility should be addressed well in advance, so families have time to select and apply to a facility, plan payment for long-term care, and make needed emotional adjustments.2
Conclusion
Although the benefits of current treatments for AD are modest, they represent a major step in the pharmacotherapy of the disease. Modest improvements provide hope to patients, caregivers, and health care providers. The identification of compounds that lower beta-amyloid burden without affecting other important physiologic pathways might represent an unequalled start in the discovery of new chemical entities for the treatment of AD. Patients who are diagnosed and treated early with medications that stabilize cholinergic function and prevent cholinergic neuron death may live to see these disease-modifying approaches.
Due to the multiple interactive mechanisms involved in the pathophysiology of AD, a multifaceted treatment strategy--in which patients receive a combination of pharmacotherapeutic agents aimed at many divergent yet overlapping pathways involved in AD--may prove to be the most beneficial. It is likely that multiple classes of medications will be useful in the treatment of AD.
Pharmacists are frequently encountered health care professionals, and opportunities for patient involvement are endless. Communicating with patients and the health care team is the first step to active involvement in patient outcomes. Pharmacists can also provide patients with the necessary information to make informed choices regarding caregiver support for patients with AD and to share realistic expectations of the available medications for the treatment of the cognitive symptoms of AD. An attitude of support and encouragement from pharmacists helps ensure the continued advancement of the pharmacy profession.
References
1. Gasparini L, Ongini E, Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer's disease: old and new mechanisms of action. J Neurochem. 2004;91:521-536.
2. Practice guideline for the treatment of patients with Alzheimer's disease and other dementias of late life. American Psychiatric Association. Am J Psychiatry. 1997;154(5 suppl):1-39.
3. Standridge JB. Pharmacotherapeutic approaches to the treatment of Alzheimer's disease. Clin Ther. 2004;26:615-630.
4. Caballero J, Nahata M. Do statins slow down Alzheimer's disease? A review. J Clin Pharm Ther. 2004;29:209-213.
5. Brower V. Harnessing the immune system to battle Alzheimer's: some of the most promising approaches to fight Alzheimer's disease aim to develop vaccines. EMBO Rep. 2002;3:207-209.
6. Potyk D. Treatments for Alzheimer disease. South Med J. 2005;98:628-635.
7. Szekely CA, Thorne JE, Zandi PP, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159-169.
8. Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke. 2004;35:2620-2622.
9. Norfray JF, Provenzale JM. Alzheimer's disease: neuropathologic findings and recent advances in imaging. Am J Roentgenol. 2004;182:3-13.
10. Farlow MR. NMDA receptor antagonists: a new therapeutic approach for Alzheimer's disease. Geriatrics. 2004;59:22-27.
11. Schmitt B, Bernhardt T, Moeller HJ, et al. Combination therapy in Alzheimer's disease: a review of current evidence. CNS Drugs. 2004;18:827-844.
12.
13. Zamrini E, De Santi S, Tolar M. Imaging is superior to cognitive testing for early diagnosis of Alzheimer's disease. Neurobiol Aging. 2004;25:685-691.
14. Rabins PV. Guideline watch: practice guideline for the treatment of patients with Alzheimer's disease and other dementias of late life. Available at: www.psych.org/psych_pract/treatg/pg/Alzheimers.watch.pdf. Accessed March 10, 2007.
15. Folstein MF, Folstein SE, McHugh PR. "
16. Hsiung GY. Current pharmacological management of Alzheimer's disease and vascular dementia. Geriatr Aging. 2006;9:22-28.
17. Cummings JL. Alzheimer's disease. N Engl J Med. 2004;351:56-67.
18. Johannsen P. Long-term cholinesterase inhibitor treatment of Alzheimer's disease. CNS Drugs. 2004;18:757-768.
19. Whitehead A, Perdomo C,
20. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer's disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317-324.
21. Berman K, Brodaty H. Tocopherol (vitamin E) in Alzheimer's disease and other neurodegenerative disorders. CNS Drugs. 2004;18:807-825.
22. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336:1216-1222.
23. Le
24 Kanowski S, Herrmann WM, Stephan K, et al. Proof of efficacy of the ginkgo biloba special extract EGb 761 in outpatients suffering from mild to moderate primary degenerative dementia of the Alzheimer type or multi-infarct dementia. Pharmacopsychiatry. 1996;29:47-56.
25. Brinton RD. Impact of estrogen therapy on Alzheimer's Disease. CNS Drugs. 2004;18:405-422.
26. Walker LC, Ibegbu CC, Todd CW, et al. Emerging prospects for the disease-modifying treatment of Alzheimer's disease. Biochem Pharmacol. 2005;69:1001-1008.
27. Robinson SR, Bishop GM, Lee HG, Munch G. Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging. 2004;25:609-615.
28. Morgan D, Gitter BD. Evidence supporting a role for anti-Abeta antibodies in the treatment of Alzheimer's disease. Neurobiol Aging. 2004;25:605-608.
To comment on this article, contact editor@uspharmacist.com.


