US Pharm. 2015;40(7):45-49.
ABSTRACT: Cystic fibrosis (CF) is a complex disease state affecting a variety of organs and body functions as the result of faulty chloride secretion caused by a genetic mutation. Complications involving the lungs, liver, and pancreas create challenging issues related to pharmacotherapy. Current advances have made dramatic improvements in patient quality of life and life expectancy. Pharmacists play a vital role in managing complex pharmacotherapy regimens around unique pharmacokinetic profiles. Novel formulations of medications and unusual sites of drug administration make the pharmacist a central provider for proper CF care.
Cystic fibrosis (CF) is the most common life-threatening genetic disorder of the Caucasian population. This disease affects almost 30,000 children and adults in the United States, with about 1,000 new cases diagnosed every year.1 Complications are caused by an autosomal recessive genetic mutation in the CF transmembrane conductance regulator (CFTR) gene located on chromosome 7, resulting in alterations of CFTR protein function. CFTR proteins act as chloride ion channels usually located on the apical portion of epithelial cells in the airway, pancreas, intestines, sweat glands, and vas deferens. Absent or decreased chloride secretion causes patients to produce thick, viscous mucous secretions that are difficult or impossible to clear. Infections, inflammation, and progressive lung deterioration are classic sequelae of this condition.1-3
Pathophysiology and Diagnosis
Due to advances in therapy, the life expectancy of people with CF has increased. From 1992 to 2012, the predicted median survival increased from 29.4 to 41.1 years. During the same time period, the median age at diagnosis decreased from 6 months to 4 months, likely because of increased newborn screening. Earlier detection of CF prevents further functional deterioration that can occur without preventive care.2
Newborn screenings that test positive for elevated immunoreactive trypsinogen indicate possible pancreatic dysfunction and prompt DNA screening to assess for CFTR genetic mutations. The gold standard test for CF is the sweat chloride test, in which a sweat gland is stimulated with pilocarpine and the resultant sweat is weighed and analyzed for its chloride concentration. A higher than expected sweat chloride content is indicative of CF.2
Pharmacokinetic Alterations
CF patients have unique pharmacokinetic and pharmacodynamic alterations as a function of their disease state. Hypotheses related to the apparent differences in pharmacokinetics in a similar healthy patient include specific drug properties affected by organ system complications and decreased overall adipose tissue. The organ systems involved, such as the lungs, gastrointestinal (GI) tract, and liver affect absorption and distribution of many drugs, and a decreased amount of adipose tissue also alters the expected solubility and distribution of drugs. Overall total body clearance of medications is increased with a larger volume of distribution noted. Larger doses may be necessary for some medications as well as an increased frequency of administration to achieve therapeutic levels comparable to those in a similar healthy patient.4
Chronic Respiratory Complications
Respiratory complications are one of the main concerns in CF care. Viscous respiratory secretions are not adequately cleared and become reservoirs for bacterial colonization and overgrowth. Airway clearance therapy is recommended to increase clearance of respiratory secretions on a routine basis regardless of exacerbation status. Airway clearance therapy may involve manual percussions in various anatomical positions with or without the assistance of airway clearance devices. Continuation of these therapies is crucial for patients in acute exacerbations and must be individualized toward each patient’s routine.4
Active treatment against bacterial colonization and overgrowth is necessary to prevent and control CF-related respiratory exacerbations. The Cystic Fibrosis Foundation (CFF) guidelines on chronic CF respiratory care recommend inhaled tobramycin for patients aged ≥6 years who have persistent airway cultures growing Pseudomonas aeruginosa, including asymptomatic carriers, to reduce the incidence of exacerbations. Other inhaled antibiotics with activity against P aeruginosa, including colistin, gentamicin, and ceftazidime, have been found to have inconclusive evidence for routine chronic administration for prevention of exacerbations.5
Mucolytic Therapies: Several nonantibiotic agents may be utilized to control respiratory complications (TABLE 1).6 Pulmozyme (dornase alfa) is strongly recommended to improve lung function and prevent exacerbations by degrading free DNA within mucus secretions. This drug effect modifies the viscosity of the secretions and allows the patient to more easily clear the airway. Hypertonic saline should also be used chronically to prevent exacerbations by altering the viscosity of mucous secretions.5 Studies have used various concentrations, often 6% to 7% sodium chloride. Nebulized hypertonic saline may cause a reactive airway and may be used concomitantly with short-acting beta2-receptor agonists.6,7
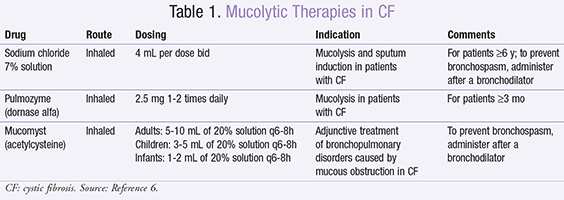
Various anti-inflammatory agents have been studied for potential benefit in improving respiratory conditions in CF. Chronic corticosteroid use, both inhaled and oral, is not recommended for management of respiratory complications due to a lack of perceived clinical benefit. Chronic use of ibuprofen is recommended to slow the deterioration of lung function; however, the drug should be used cautiously in relation to adverse effects such as possible nephrotoxicity. Chronic use of leukotriene modifiers such as montelukast or cromolyn is not recommended. For patients with airway cultures persistently positive for P aeruginosa, oral azithromycin is recommended and has been shown to be effective at preventing exacerbations of CF.5
Bronchodilators have been evaluated in chronic CF respiratory care. Inhaled short-acting beta2-adrenergic receptor agonists should be used chronically; however, inhaled anticholinergics lack the data to recommend chronic administration. Chronic use of inhaled N-acetyl-cysteine lacks sufficient evidence of efficacy for exacerbation prophylaxis.5
Chronic GI Complications
GI disorders complicate CF care because of their relationship to common disease states, but they require distinctly different pharmacotherapy. Due to the chloride channel mutation and thickened viscous secretions in CF, pancreatic and biliary ducts become obstructed, leading to hepatic and pancreatic fibrosis by retrogradation of digestive enzymes. The decreased clearance and fibrosis lead to respective enzyme deficiencies for various exocrine and endocrine functions. With the decreased amount of pancreatic enzymes secreted into the small intestine, nutritional absorption is further decreased, specifically of fat and protein along with fat-soluble vitamins.4,8
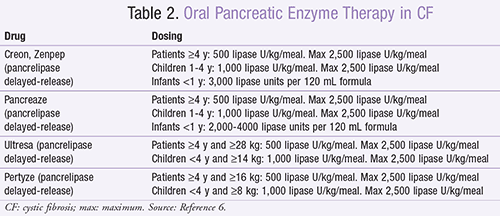
Pancreatic complications may be partially relieved by current pharmacotherapy. Pancreatic enzyme supplementation may help to decrease symptoms such as steatorrhea and abdominal pain. Usual pancreatic enzyme dosing is listed in TABLE 2.6 Titration of enzyme supplementation involves monitoring the patient’s symptom response and amount of steatorrhea. Pancreatic enzymes at doses above 10,000 U/kg/day of lipase risk fibrosing colonopathy; therefore, dosing is not recommended past this limit. The addition of antacid therapy with a histamine2 receptor blocker or a proton pump inhibitor may increase the amount of enzymes that are delivered to the small intestine for absorption by increasing the pH of the gastric acid. Patients should be counseled to not chew or crush the tablets or capsules, but they may sprinkle the capsule contents onto acidic soft food such as applesauce. Rinsing of the patient’s mouth after administration, especially in infants, protects against ulceration.4,8
A separate manifestation but also related to the pancreas, CF-related diabetes (CFRD) ultimately affects up to half of all adult patients with CF.4 CFRD is uniquely classified apart from type 1 and 2 diabetes mellitus in that effects of insulin sensitivity are transiently decreased with periods of inflammation, and beta-cell production of insulin may be decreased or delayed. Postprandial hyperglycemia is common, but ketosis is rare in most cases of CFRD.4,8,9
Liver manifestations occur in up to 70% of patients, most commonly hepatic steatosis. Liver disease usually presents around puberty and develops insidiously thereafter. Treatment involves optimizing supportive therapies such as nutrition supplementation and monitoring complications of portal hypertension. Ursodeoxycholic acid has been used for prevention of complications but with little data to show benefit.8
Nutritional requirements fluctuate based on a variety of factors but are increased overall 110% to 200% of recommended allowances. Nutritional alterations account for development of fat-soluble vitamin and micronutrient deficiencies, growth and pubertal delays, and bone disorders. Vitamin supplementation in CF patients is not well studied; however, bone disorders may be prevented with proper vitamin D and calcium supplementation in combination with physical exercise. Bisphosphonate therapy has shown benefit in adult CF patients, but data on fracture rate and survival are lacking in large longitudinal studies.10
Fertility Issues
Nearly all males with CF are born without a vas deferens, resulting in infertility. Some men with this condition who wish to father children have the option of assisted fertilization through aspiration of sperm from the testes. Women with CF also can experience fertility issues due to mucus-related obstruction of the cervix.2
Advances in nutrition and lung function have also contributed to an increased number of healthy pregnancies in women with CF. From 1997 to 2012, the number of women reported to be pregnant by the CFF Registry increased from 137 to 249 annually.1
Pulmonary Exacerbations
Pulmonary exacerbations are one of the main clinical manifestations that will affect pharmacist-provided care. The CFF publishes recommendations for management of pulmonary exacerbations, which include general guidance on the care setting. Home care may be an option for patients whose care would be no different from that received as an inpatient. One controversial area of treatment involves whether to discontinue an inhaled antibiotic if the same agent is being administered via IV. There is insufficient evidence available to conclude this practice either way.11
Another perplexing question is whether to treat Pseudomonas infections with one or multiple agents. There is insufficient evidence to determine a specific recommen-dation.11 Further unanswered questions include extended-interval aminoglycoside dosing and continuous infusions of beta-lactam antibiotics. Regarding aminoglycosides, once-daily dosing is recommended over traditional dosing because of its optimization of pharmacokinetic profiles. Once-daily dosing provides overall reduced exposure to potential nephrotoxins and utilizes the postantibiotic effect observed with aminoglycosides. While this recommendation stands on favorable data from the literature, there is no clear guidance on how precisely to monitor extended-interval aminoglycosides, particularly in pediatric populations. Understanding the pharmacokinetic profiles of amino-glycosides and applying that knowledge to the institution’s current drug delivery system is an ideal position from which pharmacists can improve patient care with optimal outcomes while reducing potential toxicities.11
Because of the altered pharmacokinetics in CF, antibiotics need to be assessed on an individual basis in regard to appropriate route, dose, and frequency. Respiratory administration of antibiotics has become a novel area for disease management. Potential systemic toxicities may be avoided by employing the local administration of nebulized medications. Specific nebulized antibiotics for CF exacerbations are listed in TABLE 3.6,12-15 Pharmacists must be aware of this option as a delivery system and address the challenges it presents with medication dose, concentration, and stability.11
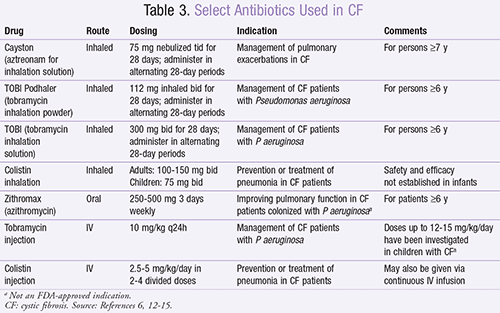
Continuous infusion of beta-lactam antibiotics has become popular at some institutions for control of CF exacerbations; however, there is currently insufficient evidence to recommend regular use. There is insufficient evidence to support duration of antibiotic therapy, but consensus in the literature indicates 14 to 21 days based on the patient’s response to therapy.11
New or Emerging Therapies
The CFF maintains on its website (www.cff.org) resources helpful in identifying new therapies under development. Recent advancements include novel formulations of aerosolized antibiotics and targeted therapy. Ivacaftor (Kalydeco) was FDA-approved in 2012 for treatment of CF in patients aged ≥6 years who have certain CFTR genetic mutations.16 Unfortunately, only 4.3% of patients with CF in 2012 had the necessary mutations to benefit from therapy.1,16 In March 2015, the drug’s indication was expanded to include children aged 2 to 5 years. Ivacaftor facilitates chloride transport by increasing the probability of opening chloride channels of the CFTR protein. The drug has resulted in major weight and lung function improvements. Patients should be counseled to take the medication with fat-containing foods such as peanut butter, cheese pizza, or whole-milk dairy products.16 Ivacaftor is the first FDA-approved drug for CF that is disease-modifying rather than addressing only the consequences of the disease, thus giving hope for future pharmacologic targets.3 Specific indications and dosing considerations can be found in TABLE 4.1,16

Role of the Pharmacist
The specific role of a pharmacist in CF management is highly specialized and integral to producing positive patient outcomes. While no specific consensus documents exist in the U.S., the United Kingdom published a document in 2011 that specifically focuses on this role.17 The consensus document maintains a principle objective of patient-focused pharmaceutical care and cooperation with other healthcare professionals in developing, implementing, and monitoring therapeutic outcomes. The document specifically cites that a dedicated CF clinical pharmacist reduces medication errors and increases availability of medications.17
In the U.S., CF patients are found in many places where clinical pharmacist services are not always available; therefore, pharmacists may be challenged with providing complex medical care to such patients at any time. Handling the pharmacokinetic and pharmacodynamic parameters, compounding unique medication formulations such as aerosolized antibiotics, and coordinating pharmaceutical care with specialty pharmacies is needed for complete and appropriate patient care.
Conclusion
CF is a serious disease state affecting a variety of organs and body functions. Pharmacists play a unique role in managing complex pharmacotherapy regimens around unique pharmacokinetic profiles. Novel formulations of medications and unusual sites of administration also make a pharmacist a central provider of proper CF care.
REFERENCES
1. Cystic Fibrosis Foundation. Patient Registry 2012 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation; 2013. www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf. Accessed June 3, 2015.
2. Montgomery GS, Howenstine M. Cystic fibrosis. Pediatr Rev. 2009;30(8):302-310.
3. O’Reilly R, Elphick HE. Development, clinical utility, and place of ivacaftor in the treatment of cystic fibrosis. Drug Des Devel Ther. 2013;7:929-937.
4. Chisholm-Burns M, Wells B, Schwinghammer T, et al. Pharmacotherapy: Principles and Practice. 3rd ed. New York, NY: McGraw-Hill Medical; 2013.
5. Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176(10):957-969.
6. Clinical Pharmacology [online database]. www.clinicalpharmacology.com. Tampa, FL: Gold Standard, Inc; 2015. Accessed March 2, 2015.
7. Taylor-Robinson DC, Schechter MS, Smyth RL. Comparing cystic fibrosis outcomes across the pond. Thorax. 2015;70(3):203-204.
8. Ledder O, Haller W, Couper RT, et al. Cystic fibrosis: an update for clinicians. Part 2: hepatobiliary and pancreatic manifestations. J Gastroenterol Hepatol. 2014;29(12): 1954-1962.
9. Bridges N. Diabetes in cystic fibrosis. Paediatr Respir Rev. 2013;14(suppl 1):16-18.
10. Haller W, Ledder O, Lewindon PJ, et al. Cystic fibrosis: an update for clinicians. Part 1: nutrition and gastrointestinal complications. J Gastroenterol Hepatol. 2014;29(7):1344-1355.
11. Flume PA, Mogayzel PJ Jr, Robinson KA, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180(9):802-808.
12. TOBI Podhaler (tobramycin) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation; November 2014.
13. Antibiotic Treatment for Cystic Fibrosis. Report of the UK Cystic Fibrosis Trust Antibiotic Working Group. 3rd ed. May 2009. www.cysticfibrosis.org.uk/media/82010/antibiotic-treatment-for-cystic-fibrosis-may-09.pdf. Accessed June 3, 2015.
14. Vic P, Ategbo S, Turck D, et al. Efficacy, tolerance, and pharmacokinetics of once daily tobramycin for pseudomonas exacerbations in cystic fibrosis. Arch Dis Child. 1998;78(6): 536-539.
15. Massie J, Cranswick N. Pharmacokinetic profile of once daily intravenous tobramycin in children with cystic fibrosis. J Paediatr Child Health. 2006;42(10):601-605.
16. Kalydeco (ivacaftor) package insert. Boston, MA: Vertex Pharmaceuticals Inc; March 2015.
17. Pharmacy Standards of Care. Report of the UK Cystic Fibrosis Pharmacists’ Group. 2nd ed. November 2011. www.cysticfibrosis.org.uk/media/82070/cd-pharmacy-standards-of-care-nov-2011.pdf. Accessed June 9, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.


