US Pharm. 2016;41(7)(Specialty & Oncology suppl):13-18.
ABSTRACT: Due to the increased life expectancy of HIV-infected patients on highly active antiretroviral therapy (HAART), there has been a decline in the incidence of AIDS-defining malignancies. However, a greater percentage of these patients are now being diagnosed with non–AIDS-defining malignancies, such as certain cancers. The current paucity of literature detailing the overlapping toxicity profiles and pharmacokinetic interactions between HAART and antineoplastic agents as well as the lack of guidelines regarding the optimal pharmacologic regimens for concurrent treatment of HIV and malignancies further complicate therapy. Potential drug interactions of combined treatment modalities in patients diagnosed with both HIV and cancer should be considered.
The administration of highly active antiretroviral therapy (HAART) in HIV-positive patients has had a significant impact on their overall survival. In fact, due to decreased mortality and increased life expectancy in HIV-positive patients on HAART, there has been a decreased incidence of AIDS-defining malignancies (ADMs) such as Kaposi’s sarcoma, non-Hodgkin’s lymphoma (NHL), and cervical cancer.1 However, there has been an increased incidence of non–AIDS-defining malignancies (NADMs) such as Hodgkin’s lymphoma (HL), anal carcinoma, lung carcinoma, head and neck cancers, and skin cancers that have been reported in the HIV-infected population at higher than expected frequencies than in the general population.1
The risk of an HIV-infected individual developing NADMs is often multifactorial and likely attributable to lifestyle; coinfection with oncogenic viruses such as human papillomavirus (HPV), hepatitis B and C, and Epstein-Barr virus; low CD4+ cell counts (<500 cells/mm3); and/or exposure to a high HIV viral load. The evidence regarding the latter two risk factors suggests that initiating HAART to both suppress HIV viral replication and maintain CD4+ cell counts >500 cells/mm3 reduces the overall incidence of ADMs and, potentially, NADMs.2,3 Based on these findings, the optimization of HAART is currently recommended in cancer patients infected with HIV.4
As 33% of all HIV-related deaths are attributable to cancer, concomitantly treating such patients with HAART and antineoplastic agents is becoming increasingly common-place.2 While the individual toxicities and drug interactions associated with both the antiretroviral and cytotoxic or molecularly targeted antineoplastic drug classes have been extensively studied, there is a clear paucity of literature on their overlapping toxicity profiles and pharmacokinetic inter-actions, not to mention the lack of national guidelines regarding the optimal therapeutic regimens for concurrent treatment of HIV and cancer.
This article will highlight the most notable pharmacologic concerns regarding the concurrent administration of antiretroviral and antineoplastic agents and provide evidence-based findings regarding agent selection and dose determination.
HAART and Chemotherapy
Several studies have found that intensive chemotherapy regimens produce similar outcomes in both HIV-infected and noninfected patients with Burkitt’s lymphoma, diffuse large B-cell lymphoma (DLBCL), and HL.5 In a study conducted in HIV-positive patients diagnosed with lung cancer, HAART use did not impact overall survival; however, cancer-specific survival was significantly higher in patients with CD4+ cell counts ≥200 cells/mm3 (an indirect effect of HAART).6 A retrospective study of patients with DLBCL being treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) and either a protease inhibitor (PI)-based regimen or non–PI-based regimen found that CHOP had a similar efficacy and toxicity profile in both antiretroviral groups.7 Lastly, in a trial conducted to analyze different antiretroviral regimens in HIV-infected patients with cancer, PI-based regimens were found to have the least favorable impact, while non-nucleoside reverse transcriptase inhibitor (NNRTI)- and integrase strand transfer inhibitor (INSTI)-based regimens had similar efficacies.8 These studies, among others, suggest that the concomitant use of antiretrovirals and chemotherapy is usually tolerable, not associated with life-threatening toxic effects, and produces response and disease-free survival rates similar to those observed in patients without cancer.
For the general population, the preferred HAART regimen includes two nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) with either an NNRTI, PI (preferably boosted with ritonavir), or an INSTI.3 Many clinicians tend to prescribe INSTI-based regimens along with chemotherapy given the considerable drug interaction and tolerability concerns rendered by the other antiretroviral classes.2 While first-line HIV regimens may be utilized in cancer patients, HAART regimens should be individualized to the chemotherapy treatment plan, baseline liver and renal function, potential for adverse events (AEs) and drug interactions, and patient preference.
There has been some controversy regarding whether or not to initiate HAART simultaneously with chemotherapy. Some of the barriers to initiating and maintaining HAART regimens in cancer patients include patients’ unwillingness to be treated for both conditions synchronously, the need for patients to be followed by multiple providers such as an oncologist and an infectious-disease specialist, and concerns for medication noncompliance and ensuing genetic variation and drug resistance.2 In nonemergent situations, the period of waiting for malignancy staging, pathology/molecular testing results, and insurance approvals should provide sufficient time to start HAART prior to initiating chemotherapy.
Several conditions that increase the urgency for commencing HAART include AIDS-defining conditions, CD4+ counts <200 cells/mm3, and rapidly declining CD4+ counts (i.e., a decrease >100 cells/mm3/year).3 ADMs and NADMs for which chemotherapy is anticipated to reduce CD4+ counts also warrant immediate HAART initiation. Furthermore, studies have found that patients with CD4+ counts 500 cells/mm3 or with opportunistic infections experienced significantly lower rates of AIDS progression and death when HAART was started early, since improvement in immune response is critical in preventing clinical progression of HIV.9,10 Alternatively, some clinicians may opt to delay HAART therapy out of concern for both overlapping toxicity profiles between HAART and chemotherapy and patient compliance.2 In such instances, clinicians may decide to add on antiretrovirals once the chemotherapy-induced AEs have been effectively managed. This methodology avoids the cycle of starting and stopping HAART, which thereby minimizes the risk for resistance. There have been studies, however, attesting to the safety and efficacy of either approach.11
A stable HAART regimen may be modified prior to initiating chemotherapy in order to reduce toxicity, improve adherence and tolerability, and avoid drug interactions.2 While antiretroviral therapy interruptions should normally be avoided, they can occur perioperatively or when antineoplastic drugs have clinically significant drug interactions with the antiretrovirals and no alternative antineoplastic medications are available. In such cases, providers should be cognizant of the differing half-lives of the antiretrovirals so as to avoid functional monotherapy, which would increase the risk of resistant mutations (i.e., efavirenz’s half-life ranges between 52 and 76 hours, which emphasizes the importance of a more staggered approach to its discontinuation). All attempts to resume antiretroviral therapy should be made when clinically appropriate. In situations when patients have poor prognostic outcomes with higher CD4+ cell counts, it may be reasonable to forgo anti-retroviral therapy completely.
With regard to the antiretrovirals, some pharmacokinetic considerations have been published, based on individual drug classes. TABLE 1 summarizes antiretroviral and chemotherapy interactions.1,2,12
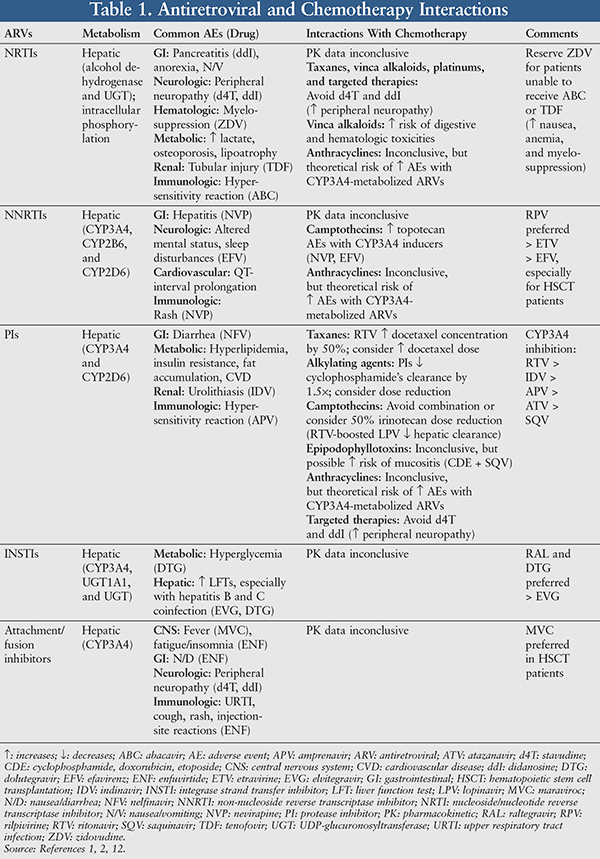
Interactions With HAART
NRTIs: As most of these agents are not eliminated by the CYP450 system, there is minimal concern for metabolism-related interactions.1 When abacavir is being used, however, screening for HLA-B*5701 should be performed to reduce the risk for hypersensitivity.2 Zidovudine and abacavir are metabolized hepatically, and didanosine, stavudine, lamivudine, and tenofovir are primarily cleared renally.1 Zidovudine commonly causes nausea, anemia, and myelosuppression, which can also be potentiated by chemotherapy, and it should be reserved for cancer patients unable to receive abacavir or tenofovir. Furthermore, since didanosine and stavudine can cause irreversible peripheral neuropathy, use of these agents should be avoided in patients on simultaneous chemotherapy commonly associated with peripheral neuropathy, such as platinum-based agents, taxanes, vinca alkaloids, and proteasome inhibitors.1,12
NNRTIs: This class of drugs undergoes extensive hepatic metabolism.1 Nevirapine is an inducer of both CYP3A4 and CYP2B6, while efavirenz can both inhibit and induce CYP3A4, in addition to inducing CYP2B6. These HAART-induced changes to homeostatic hepatic metabolism present potential complications upon coadministration with certain oncologic agents such as cyclophosphamide, ifosfamide, lomustine, etoposide, docetaxel, paclitaxel, vincristine, imatinib, erlotinib, bortezomib, tamoxifen, and procarbazine.13 Although there are limited data published on the second-generation NNRTIs, reports have noted that etravirine exhibits unpredictable drug interactions with immunosuppressants, which may be problematic for patients who have undergone hematopoietic stem cell transplantation (HSCT).1 Rilpivirine, alternatively, does not induce CYP450 and should, therefore, not theoretically affect immunosuppressant drug levels.
PIs: Ritonavir is the most potent CYP3A4 inhibitor of the PI class, and saquinavir is the least potent inhibitor, with indinavir, amprenavir, and atazanavir falling in-between.2 The potential for drug interactions with ritonavir is further exacerbated by its inhibition of CYP2D6 and its induction of CYP1A2. Studies have found that the concomitant administration of PIs with chemotherapy caused significant hematologic AEs, such as neutropenia and infections requiring hospitalizations, when compared to non–PI-based regimens.8,14 Additionally, since many PIs cause QTc prolongation, they should be used cautiously in patients on concurrent chemo-therapy that can prolong QTc intervals, such as anthracyclines, arsenic trioxide, tyrosine kinase inhibitors, and tamoxifen.2
INSTIs: Recent guidelines suggest that raltegravir-based regimens be considered for patients with malignancies due to raltegravir’s favorable drug interaction profile.1,2 Raltegravir undergoes glucuronidation by the UDP-glucuronosyltransferase (UGT) isoenzyme UGT1A1 and has a lower potential for drug interactions than PIs or NNRTIs.2 Elvitegravir is a CYP3A4 substrate and requires boosting with cobicistat, a potent CYP3A4 inhibitor, which may result in drug interactions with concomitant medications. Dolutegravir has the least postmarketing data thus far. As it is metabolized primarily by UGT, dolutegravir has minimal CYP3A4 metabolism and is expected to have a favorable drug interaction profile with chemotherapeutic drugs.2
Attachment/Fusion Inhibitors: Maraviroc is a CCR5 attachment inhibitor and has a theoretical advantage in patients undergoing HSCT, as the CCR5 receptor plays a role in the pathogenesis of graft-versus-host disease (GVHD).2,15 A phase I/II trial studying the effects of maraviroc in combination with standard GVHD prophylaxis (i.e., tacrolimus and methotrexate), following administration of reduced-intensity conditioning for allogeneic HSCT, found that the addition of maraviroc was associated with a low incidence of severe GVHD; there was no evidence of liver or gastrointestinal GVHD through day +100 (although skin GVHD was observed at expected rates), and there was no impact on engraftment, relapse, or infectious complications.15 Of note, maraviroc requires receptor tropism screening, is a substrate of CYP3A4 and the ABCB1 transporter, and is susceptible to many drug interactions.
Enfuvirtide is not hepatically metabolized and not expected to result in any significant drug interactions with chemo-therapeutic agents.1
Interactions With Antineoplastic Therapy
When considering the overlapping pharmacokinetic considerations of HAART and cytotoxic/antineoplastic agents, several studies and case reports have been useful in elucidating this otherwise murky area.
Taxanes: Paclitaxel is biotransformed into inactive metabolites by CYP2C8 and CYP3A4.1 The extent of its neurologic toxicity is correlated with the dose and area under the curve (AUC).2 One study investigated a dose reduction of 20% to 30% in patients with breast cancer and mild hepatic dysfunction.16 The authors noted that the paclitaxel serum concentration was twice as high in the study population as in patients with normal hepatic function, and correlated this with a greater incidence of grade 4 neutropenia. No effect on response rate was noted, however.16 Case reports on patients receiving paclitaxel with concomitant HAART regimens are conflicting with regard to pharmacokinetic interactions and the level of AEs that ensued, which further underscores the importance of vigilant monitoring in this patient population.1 Only one case report and one case series showed that there was no change in paclitaxel’s serum concentration or pharmacokinetics in patients who received concomitant PI-based HAART regimens.17,18
Docetaxel is metabolized by CYP3A4 into four or more inactive metabolites.1 While the AUC of docetaxel is not predictive of its toxicity, elevations in transaminases and serum alkaline phosphatase are predictive of the risk of neutropenia and mucositis. While no human pharmacokinetic data with docetaxel exist, ritonavir has been found to increase docetaxel plasma levels by 50-fold in the murine model.19,20
Alkylating Agents: Cyclophosphamide and ifosfamide are prodrugs that undergo extensive CYP450 metabolism by CYP2B6 and CYP3A4.1 Cyclophosphamide does not need to be dose adjusted for hepatic insufficiency, but requires dose reduction for hypoalbuminemia. Ifosfamide has not been shown to require hepatic dose adjustments either. One study found that when a PI-based regimen was combined with CHOP, cyclophosphamide’s clearance was decreased by 1.5-fold.8 Another study found that the incidence of neutropenia and infection related to cyclophosphamide, doxorubicin, and etoposide (CDE) was higher when combined with PIs than with NNRTI-based HAART regimens.21
Vinca Alkaloids: These drugs are CYP3A4 substrates.2 Case reports have documented the pharmacokinetic interactions between vinblastine and HAART regimens. One report detailed the development of profound neutropenia, severe constipation, and peripheral neuropathy after the patient’s HAART regimen (i.e., zidovudine, lamivudine, abacavir, nevirapine, and ritonavir-boosted lopinavir) was reinitiated.22 The association was confirmed when HAART was held and the patient experienced incomplete and short-lasting effects when the vinblastine was restarted at lower doses, while the full dose of vinblastine was effective and well tolerated. When HAART was resumed, the vinblastine was given at a reduced dose (i.e., a third of the original dose), which resulted in it being oncologically effective and well tolerated. The proposed mechanism of this drug interaction involved PI inhibition of the CYP450 enzyme system and modulation of P-glycoprotein inhibition, which reduced both the metabolism and efflux of vinblastine and lead to increased toxicities.
Camptothecins: Irinotecan is a prodrug that gets converted to its active metabolite, SN-38, by liver carboxylesterases, which gets further converted to SN-38 glucuronide (SN-38G) by UGT.2 Irinotecan also undergoes metabolism by CYP3A4 into inactive metabolites. One study reported that coadministration of irinotecan and ritonavir-boosted lopinavir resulted in a 47% reduction in the clearance of irinotecan and an increased availability of irinotecan for conversion to the active SN-38 metabolite.1,23 However, despite a 50% dose reduction, one patient needed to stop treatment with irinotecan due to persistent grade 2 neutropenia.24 Topotecan is primarily metabolized into an inactive compound via enzymatic hydrolysis, and it undergoes minor N-demethylation by the CYP450 system to form an active metabolite. While topotecan should not theoretically exhibit drug interactions with CYP3A4 inhibitors, CYP3A4 inducers such as nevirapine and efavirenz may play a more clinically significant role, as they would increase the active metabolite of topotecan, N-desmethyl topotecan.1
Epipodophyllotoxins: Both etoposide and teniposide undergo CYP3A4-dependent O-demethylation, and etoposide is minimally metabolized by CYP1A2 and CYP2E1.1 The former metabolic pathway generates epipodophyllotoxin catechol and quinone metabolites, which could damage DNA. Although there have not been any pharmacokinetic studies on epipodophyllotoxins and CYP3A4-inducing or -inhibiting antiretrovirals, there are published data regarding the overlapping toxicity profile of such agents.1 One study demonstrated that more NHL patients treated with CDE and saquinavir had developed severe mucositis than those patients not treated with saquinavir.25 In contrast, another trial found that NHL patients treated after the advent of HAART exhibited less chemotherapy-associated toxicities and experienced improved survival.11
Anthracyclines: Doxorubicin is primarily metabolized via aldoketo reductase (AKR) to an inactive metabolite, doxorubicinol, which is subsequently metabolized by NADPH-dependent reductase to noncytotoxic aglycones.1 Since the CYP450 system is primarily involved in the formation of cardiotoxic free-radical species, drug interactions with CYP450 metabolized antiretrovirals are of significant clinical importance. Two pharmacokinetic studies, however, found that CHOP regimens did not impact the clearance of doxorubicin, even with concurrent exposure to PI-based HAART regimens.26,27 Another trial investigated the pharmacokinetics of liposomal daunorubicin when administered to HIV-positive Kaposi’s sarcoma patients being treated with PI-based HAART regimens; the authors did not note any significant differences in the pharmacokinetic parameters of liposomal daunorubicin and, therefore, did not recommend dose adjustments in the studied patient population.1,28
Targeted Therapies
Monoclonal antibodies, such as cetuximab, rituximab, and bevacizumab, are eliminated by the spleen and not hepatically metabolized; thus, they should not display drug interactions with CYP450 metabolized antiretrovirals.1 Conversely, epidermal growth factor receptor (EGFR) inhibitors, such as gefitinib and erlotinib, are metabolized by CYP3A4. Additionally, imatinib, sunitinib, and bortezomib (in addition to the other members of those respective drug classes) are also metabolized by CYP3A4; however, there are no currently published pharmacokinetic data regarding the combined usage of these agents with antiretrovirals. Attentiveness to the overlapping AEs between these agents is highly recommended, in addition to appropriate dose modifications, based on clinical parameters.1
As previously mentioned, there is no guidance for dose adjustments for either HAART or chemotherapy when used concurrently, which is partly due to HIV-positive patients being excluded from oncologic drug trials.12 In 2006, however, the Cancer Therapy Evaluation Program of the National Cancer Institute (NCI) issued a statement advising clinical investigators not to arbitrarily exclude HIV-positive patients from treatment trials without scientific justification.29 In line with this progressive approach to inclusion criteria for clinical trials, the AIDS Malignancy Consortium, an NCI-supported clinical trials group, has begun to address some of the issues regarding pharmacokinetic concerns with molecularly targeted agents by undertaking prospective clinical trials that focus on patients receiving these agents who are also on HAART.30
Conclusion
The increased life expectancy for patients with HIV coupled with the increasing incidence of NADMs in this select population has made the understanding of pharmacokinetic interactions between antiretrovirals and cytotoxic medications imperative. An interdisciplinary collaboration among oncologists, infectious disease specialists, and pharmacists is crucial for the determination of the safest concurrent chemotherapy and HIV regimens. Since there is currently a dearth of authoritative guides as to the selection and dosing of concomitantly administered antineoplastic and HAART regimens, much vigilance should be placed into selecting an optimal treatment plan, especially with drugs that are reliant upon hepatic enzymes for bioactivation and metabolism. In this manner, we can avoid compromising therapeutic efficacy and/or inciting inadvertent toxicities.
The present paucity of pharmacokinetic evidence regarding safe and effective therapeutic approaches in HIV-positive patients with comorbid malignancies obviates the need for further prospective trials to be conducted. The results of new studies are being eagerly anticipated by the scientific community in the hopes that they will provide much needed guidance in this clinically hazy area.
REFERENCES
1. Mounier N, Katlama C, Costagliola D, et al. Drug interactions between antineoplastic and antiretroviral therapies: implications and management for clinical practice. Crit Rev Oncol Hematol. 2009;72(1):10-20.
2. Torres HA, Mulanovich V. Management of HIV infection in patients with cancer receiving chemotherapy. Clin Infect Disease. 2014;59(1):106-114.
3. DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1 infected adults and adolescents. Department of Health and Human Services. January 28, 2016. https://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. Accessed May 31, 2016.
4. Kaplan JE, Benson C, Holmes KK, et al. Guidelines for the prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and HIV Medicine Association of the Infectious Disease Society of America. MMWR Recomm Rep. 2009;58(RR-4):1-207.
5. Montoto S, Shaw K, Okosun J, et al. HIV status does not influence outcome in patients with classical Hodgkin lymphoma treated with chemotherapy using doxorubicin, bleomycin, vinblastine, and dacarbazine in the highly active antiretroviral therapy era. J Clin Oncol. 2012;30(33):4111-4116.
6. Pakkala S, Chen Z, Rimland D, et al. Human immunodeficiency virus-associated lung cancer in the era of highly active antiretroviral therapy. Cancer. 2012;118(1):164-172.
7. Wong AY, Marcotte S, Laroche M, et al. Safety and efficacy of CHOP for treatment of diffuse large B-cell lymphoma with different combination antiretroviral therapy regimens: SCULPT study. Antivir Ther. 2013;18(5):699-707.
8. Torres HA, Rallapalli V, Saxena A, et al. Efficacy and safety of antiretrovirals in HIV-infected patients with cancer. Clin Microbiol Infect. 2014;20(10):O672-O679.
9. Zolopa A, Andersen J, Powderly W, et al. Early antiretroviral therapy reduces AIDS progression/death in individuals with acute opportunistic infections: a multicenter randomized strategy trial. PLoS One. 2009;4(5):e5575.
10. Thompson MA, Aberg JA, Hoy JF, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-US panel. JAMA. 2012;308(4):387-402.
11. Little RF, Pittaluga S, Grant N, et al. Highly effective treatment of acquired immunodeficiency syndrome-related lymphoma with dose-adjusted EPOCH: impact of antiretroviral therapy suspension and tumor biology. Blood. 2003;101(12):4653-4659.
12. Rudek MA, Flexner C, Ambinder RF. Use of antineoplastic agents in patients with cancer who have HIV/AIDS. Lancet Oncol. 2011;12:905-912.
13. Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679-1691.
14. Bower M, Powles T, Stebbing J, et al. Potential antiretroviral interactions with cyclophosphamide, doxorubicin, and etoposide. J Clin Oncol. 2005;23:1328-1329.
15. Reshef R, Luger SM, Hexner EO, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367(2):135-145.
16. Seidman AD, Hochhauser D, Gollub M, et al. Ninety-six hour paclitaxel infusion after progression during short taxane exposure: a phase II pharmacokinetic and pharmacodynamics study in metastatic breast cancer. J Clin Oncol. 1996;14(6):1877-1884.
17. Nannan Panday VR, Hoetelmans RM, van Heeswijk RP, et al. Paclitaxel in the treatment of human immunodeficiency virus 1-associated Kaposi’s sarcoma-drug-drug interactions with protease inhibitors and a nonnucleoside reverse transcriptase inhibitor: a case report study. Cancer Chemother Pharmacol. 1999;43(6):516-519.
18. Duchin K, Sun J, Tan M, et al. Pharmacokinetics of low-dose paxene (paclitaxel) in patients with refractory or relapsed AIDS-related Kaposi’s sarcoma. Proc Am Soc Clin Oncol. 1997;A829 [Abstract].
19. Bower M, McCall-Peat N, Ryan N, et al. Protease inhibitors potentiate chemotherapy-induced neutropenia. Blood. 2004;104(9):2943-2946.
20. Bardelmeijer HA, Ouwehand M, Buckle T, et al. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002;62(21):6158-6164.
21. Makinson A, Martelli N, Peyriere H, et al. Profound neutropenia resulting from interaction between antiretroviral therapy and vinblastine in a patient with HIV-associated Hodgkin’s disease. Eur J Haematol. 2007;78(4):358-360.
22. Kotb R, Vincent I, Dulioust A, et al. Life-threatening interaction between antiretroviral therapy and vinblastine in HIV-associated multicentric Castleman’s disease. Eur J Haematol. 2006;76:269-271.
23. Carona G, Vaccer E, Sandron S, et al. Lopinavir-ritonavir dramatically affects the pharmacokinetics of irinotecan in HIV patients with Kaposi’s sarcoma. Clin Pharmacol Ther. 2008;83:601-606.
24. Corona G, Vaccher E, Cattarossi G, et al. Potential hazard of pharmacokinetic interactions between lopinavir-ritonavir protease inhibitors and irenotecan. AIDS. 2005;19(17):2043-2044.
25. Sparano JA, Wiernik PH, HU X, et al. Saquinavir enhances the mucosal toxicity of infusional cyclophosphamide, doxorubicin, and etoposide in patients with HIV-associated non-Hodgkin’s lymphoma. Med Oncol. 1998;15(1):50-57.
26. Toffoli G, Corona G, Cattarossi G, et al. Effect of highly active antiretroviral therapy (HAART) on pharmacokinetics and pharmacodynamics of doxorubicin in patients with HIV-associated non-Hodgkin’s lymphoma. Ann Oncol. 2004;15(12):1805-1809.
27. Ratner L, Lee J, Tang S, et al. Chemotherapy for human immunodeficiency virus-associated non-Hodgkin’s lymphoma in combination with highly active antiretroviral therapy. J Clin Oncol. 2001;19:2171-2178.
28. Fumagalli L, Zucchetti M, Parisi I, et al. The pharmacokinetics of liposomal encapsulated daunorubicin are not modified by HAART in patients with HIV-associated Kaposi’s sarcoma. Cancer Chemother Pharmacol. 2000;45(6):495-501.
29. National Cancer Institute. Guidelines regarding the inclusion of cancer survivors and HIV-positive individuals on clinical trials. Updated May 29, 2008. http://ctep.cancer.gov/protocolDevelopment/policies_hiv.htm. Accessed September 10, 2015.
30. Deeken JF, Mitsuyasu RT, Little RF, et al. Treating HIV+ patients for non-AIDS-defining cancers (NADCs) in the era of targeted chemo-therapy: an AIDS malignancy consortium study of sunitinib in patients on ART. J Clin Oncol. 2010;28(suppl 15):TPS161.
To comment on this article, contact rdavidson@uspharmacist.com.


