US Pharm. 2018;43(5)16-21.
ABSTRACT: Cystic fibrosis (CF) is a genetic disorder that affects various body systems, leading to premature death. Newborn screening in all states has helped identify those who have this disorder and allows for earlier interventions. Although traditionally viewed as a pulmonary disorder, treatments are available to address the various manifestations of CF. Airway clearance, nutritional support, pancreatic-enzyme replacement, and chronic antibiotic therapy are among the current recommended treatments. The most recent development involves the use of cystic fibrosis transmembrane conductance regulators that help to correct the underlying problem rather than just treat the symptoms. Pharmacists can serve an important role in the proper treatment of primary and secondary symptoms of CF.
Cystic fibrosis (CF) is an autosomal recessive disease stemming from the mutation of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Although over 2,000 CFTR variants have been discovered, not all cause CF. An individual acquires CF when a gene variant (allele) is inherited from each parent.1 CFTR variants are categorized into five classes of mutations that affect how the CFTR protein is made and, ultimately, how this disorder is treated. Class 2 mutations are the most prevalent, affecting approximately 88% of CF patients.2 The mutation of the CTFR gene results in impaired or absent chloride transport across epithelial cells. CFTR most prevalently resides in the surface of epithelial cells located in the respiratory system, digestive system, and sweat glands.3
EPIDEMIOLOGY
CF is seen in individuals of all racial and ethnic backgrounds. However, of all the genetic disorders that can cause premature death, it has the highest incidence among Caucasians of North American, European, and Australian descent.4 Approximately 30,000 people are living with CF in the United States, with 1,000 new cases diagnosed every year.5 The overall prevalence in live births in the U.S. is estimated to be approximately 1 in 4,000.6 As of 2016, 93.7% of CF patients were white, 4.6% were African American, and 3.5% were of other races.5 The life expectancy of people with CF has increased significantly over the past 20 years. The estimated average life expectancy at birth has increased from 34 years during 1991–1995 to 47.7 years during 2012–2016.5
DIAGNOSIS
Most individuals born prior to 2010 were not routinely screened. However, universal newborn screening in all 50 states and the District of Columbia was implemented by 2010, thereby helping to prevent delayed and missed diagnoses.7
Earlier diagnosis is associated with beneficial effects on nutritional status, including growth improvement (height and weight) and prevention of fat-soluble vitamin deficiency and protein malnutrition.8 At least 62.4% of new CF diagnoses in the U.S. now occur in asymptomatic or minimally symptomatic infants, with the median age of diagnosis at 4 months.5
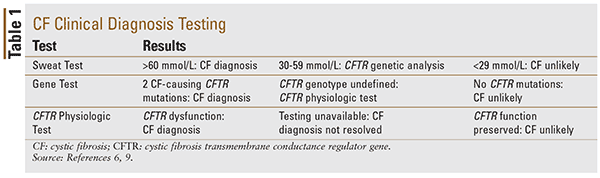
Several methods of newborn screening may be implemented to detect potential CF, such as the immunoreactivity trypsinogen test (IRT), double IRT testing, and pancreatitis-associated protein testing. A positive or equivocal screening test should be followed by CFTR genetic testing and the sweat chloride test, which involves the collection of sweat after the transdermal administration of pilocarpine in order to confirm the diagnosis.8 Sweat chloride testing is the gold standard for discriminating between true positive and false positive results in newborns.6,9 See Table 1 for CF clinical diagnostic testing.
PATHOPHYSIOLOGY
CFTR gene mutation leads to impaired or absent chloride transport across epithelial cells located in the airway, pancreas, intestines, sweat glands, and vas deferens.3 Impairment or absence of chloride transport leads to the production of thick, viscous mucus. Mucus retention in the lungs leads to chronic infection and damaging airway inflammation. Daily symptoms usually include shortness of breath and cough with sputum production. Among the causes of morbidity and mortality, the primary one is the development of progressive impairment of the respiratory system.9 Patients with CF have unique pharmacokinetic and pharmacodynamic profiles related to their disease state.
Patients with CF have an increased volume of distribution that requires larger doses of certain medications, especially antibiotics. They also have a faster rate of clearance that necessitates shorter intervals between doses.10
In addition to respiratory impairment, there may also be problems associated with the liver, pancreas, sweat glands, and vas deferens. Accumulation of viscous mucus in the pancreatic and biliary ducts may lead to pancreatic and biliary obstruction and may cause cirrhosis of the liver or portal hypertension. Pancreatic-duct obstruction can also lead to complications such as chronic obstructive pancreatitis, chronic fibrosis, and fatty replacement of the pancreatic gland.10 As patients age, the risk of CF-related diabetes mellitus increases (19% in adolescents and up to 50% of adults).11
TREATMENT
Nutritional Support
Management of CF requires good nutrition and appropriate supplementation of vitamins and pancreatic enzymes. The patient’s underlying gene mutation, increased effort of breathing, and the metabolic consequences of chronic infection require increased nutritional intake. The recommended increase in energy intake ranges from 110% to 200% of that needed for healthy individuals.12 Improved pulmonary function and survival is associated with normal weight ranges in CF patients.12 It is recommended that patients with pancreatic insufficiency consume a high-calorie diet with appropriate unrestricted fat intake. High-fat diets (40% of total calories) promote growth and improve lung function.13
Pancreatic enzyme replacement therapy (PERT) and fat-soluble vitamin supplementation are used to treat pancreatic insufficiency and improve fat absorption. Because of the inability to absorb fat-soluble vitamins A, D, E, and K, patients with pancreatic insufficiency should be monitored and given prophylactic supplementation of fat-soluble vitamins. The current recommendation for PERT from the American Society for Parenteral and Enteral Nutrition is to start with 500 lipase units per kilogram of body weight per meal up to a maximum of 2,500 units/kg/meal or 10,000 units/kg/day.13 The Cystic Fibrosis Foundation guidelines recommend starting with a low dose of 500 units per gram of dietary fat ingested per day up to 4,000 units per gram.14 The patient’s clinical response should be used to determine the dosing titration regardless of which recommendation or guideline is followed.
Pancreatic enzyme products utilize enteric-coating dosage forms because of their sensitivity to an acidic environment. Histamine-2 receptor antagonists and proton pump inhibitors are sometimes used to improve absorption of pancreatic enzymes by maintaining an alkaline pH, especially when the enzyme dosage reaches maximum levels.10,14 However, the current evidence concerning efficacy is equivocal.15,16 Patients should be counseled not to crush or chew pancreatic-enzyme capsules. The capsules may be opened and sprinkled on soft, nonalkaline foods such as apple sauce.13 A mouth rinse after use helps to prevent ulceration. Pancreatic enzymes should be administered prior to consuming meals, snacks, and fat-soluble vitamins. Patients receiving dosages above these guidelines may develop fibrosing colonopathy.10,13,14
Pulmonary Therapy: Nonpharmacologic Treatment
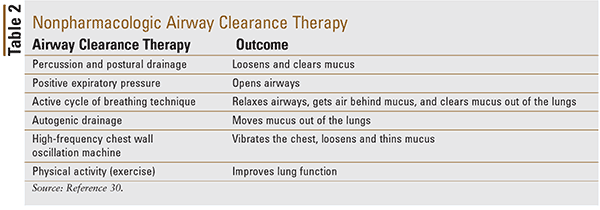
Airway clearance therapy is performed on a daily basis to improve clearance of mucus from the lungs. Postural percussion therapy or chest physiotherapy devices are routinely used by patients to loosen and clear mucus. Positive expiratory pressure devices help keep the patient’s airway open. Breathing out with moderate force through resistance allows airflow to get beneath mucus obstruction so that it can be coughed out.17 See Table 2 for a detailed list of airway-clearance therapies.
Pharmacologic Treatment
Drugs to treat CF are designed to improve the clearance of mucus from the lungs and to treat persistent infections. The most recent guidelines from the Cystic Fibrosis Foundation regarding medication use were published in 2013.18 Previous guidelines were updated and two new medications were evaluated and graded using the United States Preventive Services Task Force definitions.19
The Cystic Fibrosis Foundation recommends the following treatments as having a high certainty of substantial net benefit, grade A, for moderate-to-severe disease: inhaled tobramycin, dornase alfa, ivacaftor, and inhaled aztreonam.18 See Table 3 for a complete list of drug products with a high/substantial or high/moderate grade recommendation.
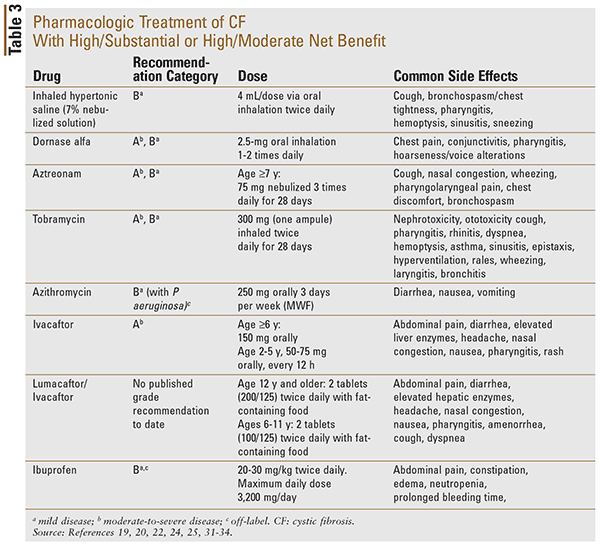
Hypertonic Saline: Inhaled hypertonic saline is administered by nebulization to restore airway hydration, induce expectoration of sputum, and enhance mucociliary function. Although 7% is the standard strength, it can be administered in a strength as low as 3% for patients experiencing side effects, which may include cough, sore throat, and chest tightness.20,21 Chronic use of inhaled hypertonic saline is recommended for patients aged 6 years and older for improved lung function and quality of life, and reduced exacerbations.18
Dornase alfa (Pulmozyme): This is a recombinant human deoxyribonuclease. It cleaves to the extracellular DNA of mucus and decreases the viscosity of mucus, making it easier to cough up. This medication is administered by nebulization route one to two times daily.22 For patients aged 6 years and older with moderate-to-severe disease, it has been shown to reduce exacerbations while also improving lung function and quality of life.18
Antibiotics: Bacterial colonization and growth in the lungs often causes pulmonary exacerbations in CF patients. Common characteristics of pulmonary exacerbations include increased coughing, diminished pulmonary function, and an increase in mucus production with a change in color.10 Acute exacerbation treatment frequently requires inpatient hospital treatment unless the home can provide a level of IV care equal to a hospital setting.23 Patients experiencing an acute exacerbation should stay on their chronic therapy along with an intensified airway clearance treatment.23 Evidence is lacking to recommend using one versus multiple antibiotics, or the optimal duration of treatment.23
In patients (aged 6 years) with chronic presence of Pseudomonas aeruginosa (P aeruginosa) in airway cultures, prophylactic use of antibiotics (tobramycin, aztreonam, and azithromycin) is recommended.18 The use of inhaled tobramycin and aztreonam delivers the drug locally to the lung and decreases the risk of systemic side effects.
For chronic use, tobramycin is administered by nebulization (300 mg) or dry powder inhalation (112 mg) twice daily for 28 days on and 28 days off.24 It can also be administered IV or IM at a dose of 10 mg/kg/day given in four equally divided doses.24 However, these routes of administration are not included in the guidelines for chronic use.18
Aztreonam inhalation solution is an alternative antibiotic for patients with chronic colonization of P aeruginosa. Although indicated for this use, it is considered to be an orphan drug for this purpose.25 Aztreonam 75 mg is administered by nebulization route three times daily for 28 days on and then 28 days off. If a patient is chronically using a bronchodilator and mucolytic, the bronchodilator should be used first, followed by the mucolytic, and then the aztreonam.25 In a head-to-head clinical trial, aztreonam inhalation solution was compared with inhaled tobramycin and demonstrated clinical superiority in improving lung function.26 Although approved for IV use from age 9 months, it is not included in the guidelines for chronic use.18
Azithromycin is an oral agent recommended by the guidelines for use with chronic P aeruginosa to improve lung function and reduce exacerbations. However, it is considered to have a slightly lower estimated end benefit when compared with inhaled tobramycin and aztreonam.18
Evidence for the chronic use of other antibiotics is inconclusive regarding improvement in lung function and quality of life, and reduced exacerbations.18
CFTR Modulator Therapies: In a recently published guideline on CFTR modulator therapy, 30 different treatment recommendations were made based on gene mutation.27 Ivacaftor (Kalydeco) was the first approved disease-modifying (potentiator) drug for CF (2012) compared with the previous drugs that only addressed the treatment of symptoms. In one study, over a 48-week period it was reported to improve lung function along with several other CF-related problems.28 Ivacaftor is approved to treat a total of 38 CFTR gene mutations. The Cystic Fibrosis Foundation guidelines recommend chronic use of ivacaftor in patients aged 2 years and older with at least one CFTR mutation for improvement in lung function and quality of life, and reduced exacerbations.27 It is available in tablet and oral granules formulations. A disadvantage of ivacaftor is that it only targets a small portion (5%) of CF patients with a G551D mutation.28
Lumacaftor (200 mg) + ivacaftor (125 mg), Orkambi, is the first approved (2015) CFTR corrector and potentiator combination therapy. It is approved for use in CF patients with two copies of the Phe508del (formerly F508del) CFTR mutation who are aged 6 years and older. Approximately 45% of the CF population in the U.S. could benefit from this therapy.29 Orkambi showed improvement in pulmonary function and reduction in pulmonary exacerbation compared with placebo in two phase III clinical trials.29 Patients should be counseled to take CFTR modulators with fat-containing food.
Anti-inflammatory Medications: The Cystic Fibrosis Foundation recommends against the chronic use of inhaled or oral corticosteroids and leukotriene modifiers owing to insufficient evidence that they can improve lung function and quality of life, or reduce exacerbations.18 Chronic use of oral high-dose ibuprofen to reach a peak plasma concentration of 50-100 mcg/mL is recommended for individuals aged 6 to 17 years with an FEV1 greater than 60% predicted in order to slow the progressive loss of lung function in this group.18 There is insufficient evidence for use of ibuprofen in patients aged 18 years or older.18
Other Chronic Treatments: There are several potential chronic medications that have neither a positive nor negative recommendation for use in patients aged at least 6 years owing to lack of evidence, according to the Cystic Fibrosis Foundation guidelines. Examples of such chronic medications include inhaled beta2 adrenergic agonists, leukotriene modifiers, inhaled or oral N-acetylcysteine or inhaled glutathione, and inhaled anticholinergics.18
Pharmacists’ Role
Universal newborn screening, the growth of information concerning CF, the refinement of treatment guidelines, and the development of new therapies are providing more and better opportunities for pharmacists to be involved in their CF patients’ care. Pharmacists need to understand the pathophysiology of this disease and the current guidelines for treatment in order to provide appropriate treatment recommendations. With the complexity of drug treatments, pharmacists need to be diligent in their performance of drug utilization reviews, regardless of practice type. Patient counseling is more important than it has ever been regarding the treatment of CF. Pharmacists can provide their patients with the most up-to-date information about their drug therapy, which can help to maximize adherence.
REFERENCES
1. Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45-56.
2. Wang Y, Wrennall JA, Cai Z, et al. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol. 2014;52:47-57.
3. Sorcher EJ. Cystic Fibrosis. Kasper D, Fauci A, Hauser S, et al. eds. In: Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
4. Bepari KK, Malakar AK, Paul P, et al. Allele frequency for Cystic fibrosis in Indians vis-a-vis global populations. Bioinformation. 2015;11(7):348-352.
5. Cystic Fibrosis Foundation Patient Registry. 2016 Annual Data Report. Bethesda, Maryland. www.cff.org/Research/Researcher-Resources/Patient-Registry/2016-Patient-Registry-Annual-Data-Report.pdf. Accessed March 30, 2018.
6. Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181S:S4-S15.
7. Cystic Fibrosis Foundation. All fifty states to screen newborns for cystic fibrosis by 2010. July 7, 2009. www.cff.org/About-Us/Media-Center/Press-Releases/All-Fifty-States-to-Screen-Newborns-for-Cystic-Fibrosis-by-2010/. Accessed April 19, 2018.
8. Castellani C. Southern KW, Brownlee K, et al. European best practice guidelines for cystic fibrosis neonatal screening. J Cyst Fibros. 2009;8:153-173.
9. Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519-2531.
10. Wright CC, Vera YY. Cystic fibrosis. In: DiPiro JT, Talbert RL, Yee GC, et al. eds. Pharmacotherapy: A Pathophysiologic Approach, 10th ed. New York, NY: McGraw-Hill; 2016.
11. Moran A, Dunitz J, Nathan B, et al. Cystic fibrosis–related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32(9):1626-1631.
12. Stallings VA, Stark LJ, Robinson KA, et al. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108(5):832-839.
13. Matel, JL. Nutritional management of cystic fibrosis. JPEN. January 2012;36 (Suppl 1):60S-67S.
14. Schwarzenberg SG, Dorsey J. Pancreatic Enzymes Clinical Care Guidelines. Bethesda, MD: Cystic Fibrosis Foundation; 2013. www.cff.org/Care/Clinical-Care-Guidelines/Nutrition-and-GI-Clinical-Care-Guidelines/Pancreatic-Enzymes-Clinical-Care-Guidelines. Accessed November 2, 2017.
15. Ng SM, Jones AP. Drug therapies for reducing gastric acidity in people with cystic fibrosis. Cochrane Database Syst Rev. 2003;2:CD003424.
16. Francisco MP, Wagner MH, Sherman JM, et al. Ranitidine and omeprazole as adjuvant therapy to pancrelipase to improve fat absorption in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35:79-83.
17. McIlwaine M, Button B, Dwan K. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst Rev. 2015;6:CD003147.
18. Mogayzel PJ, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680-689.
19. Grade definitions (July 2012). U.S. Preventive Services Task Force.
www.uspreventiveservicestaskforce.org/Page/Name/grade-definitions. Accessed March 14, 2018.
20. Sodium chloride. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
21. Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006. 354(3):229-240.
22. Dornase alfa. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
23. Flume PA, Mogayzel PJ, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802-808.
24. Tobramycin. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
25. Aztreonam. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
26. Assael BM, Pressler T, Bilton D, et al. Inhaled aztreonam lysine vs. inhaled tobramycin in cystic fibrosis: a comparative efficacy trial. J Cyst Fibros. 2012;12(2):130-140.
27. Ren CL, Morgan RL, Oermann C, et al. Cystic Fibrosis Foundation Pulmonary Guidelines: use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc. 2018;15(3):271-280.
28. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
29. Wainwright CE, Elborn JS, Ramsey BW, et al. (2015). Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. New Engl J Med; 2015;373(3):220-231.
30. Flume PA, Robinson KA, O’Sullivan BP, et. al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54(4):522-537.
31. Azithromycin. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
32. Ivacaftor. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
33. Lumacaftor; Ivacaftor. In: Clinical Pharmacology [online database]. Tampa, FL: Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
34. Ibuprofen. In: Clinical Pharmacology [online database]. Tampa, FL:
Gold Standard; 2018. www.clinicalpharmacology.com. Accessed March 14, 2018.
To comment on this article, contact rdavidson@uspharmacist.com.


