US Pharm.
2008;33(6):HS10-HS18.
Heart failure (HF) afflicts
5.3 million people in the United States, with 550,000 new cases each year.
1,2 The current treatment of HF consists of ACE inhibitors, angiotensin
receptor blockers, beta-blockers, aldosterone antagonists, digoxin, and
diuretics. Because of decreased cardiac output, patients with HF have
increased activity of the sympathetic nervous system and renin-angiotensin
aldosterone system (RAAS), along with an increase in nonosmotic vasopressin
release. Combined, this leads to diminished renal sodium and water excretion,
as well as edema and symptoms of volume overload.3 Diuretics have
been used in the treatment of patients with HF since 1919, with the discovery
of the diuretic trait of mercury.4 Currently, loop diuretics,
thiazide diuretics, and aldosterone antagonists are commonly used in patients
with HF to remove excess volume and relieve symptoms.
Epidemiology
Heart failure
contributes to approximately 300,000 deaths each year.1,2 In
addition, HF is the most common diagnosis in hospitalized patients aged 65
years and older.2 It is more prevalent in African Americans and in
people who are overweight or obese.2 Men have a higher rate of HF
than women, and 80% of men and 70% of women under the age of 65 who have HF
will die within eight years.1,2 The total cost (direct and
indirect) of HF in the U.S. for 2008 is estimated at $34.8 billion.1
Etiology and Clinical
Presentation
Intrinsic disease
states such as dilated or hypertrophic cardiomyopathy, in addition to various
external factors including uncontrolled hypertension, increased stroke volume,
and hormonal disorders, place high demands upon the heart, leading to HF.
Within the past three decades, considerable attention has focused on left
ventricular (LV) dysfunction, loading conditions, neuroendocrine activation,
and ventricular remodeling as the principal pathophysiologic mechanisms
underlying HF progression.5 There has been a fundamental
shift, however, in the origin of HF that is often underemphasized.5-7
The Framingham Heart Study suggests that the most common cause of
HF is no longer hypertension or valvular heart disease, as it was
in previous decades, but rather coronary artery disease.5 Coronary
artery disease leads to reduction in coronary blood flow and oxygen delivery
to the myocardium, resulting in hypoxia and impaired cardiac function.
Conditions such as arrhythmias, congenital defects, and cardiomyopathies of
known origins (i.e., bacterial or viral) also precipitate HF.
The cardinal manifestations of
HF vary depending on whether right ventricular (RV) or LV failure is present.
Although both types may occur together, the symptoms of one side often
predominate. Systemic edema including ascites, increased jugular venous
pressures, hepatosplenomegaly, peripheral edema, and anasarca are mainly
associated with RV failure (cor pulmonale). Dyspnea, orthopnea, paroxysmal
nocturnal dyspnea, and nocturia, all of which are related to pulmonary
congestion symptoms, are associated with LV failure.8
The approach first developed
by the New York Heart Association (NYHA) to quantify clinical assessment of HF
remains widely utilized and is described in TABLE 1.9

Pharmacologic Treatments
Management of HF
commences with a careful assessment of etiology and severity of the disease
followed by correction of systemic factors (thyroid dysfunction, infection,
etc.), lifestyle modifications (e.g., lower salt intake, alcohol cessation,
medication compliance), and a review of drugs that may contribute to HF (e.g.,
NSAIDs, antiarrhythmics, calcium channel blockers, thiazolidinediones).10
Reducing pulmonary and/or
systemic congestion along with edema and other clinical symptoms such as
dyspnea and shortness of breath is the primary goal of diuretic therapy.
Diuretic agents promote an increase in urine output by altering how the kidney
handles sodium and water, since sodium excretion is followed by water
excretion. Nonetheless, inhibition of sodium reabsorption may take place at
different segments of the renal tubular system. A synergistic effect may be
seen when using two different diuretic agents that alter sodium reabsorption
at multiple nephron sites, therefore enhancing efficacy. Properties of
the pharmacologic diuretic agents mainly utilized in treating HF are described
in TABLE 2.
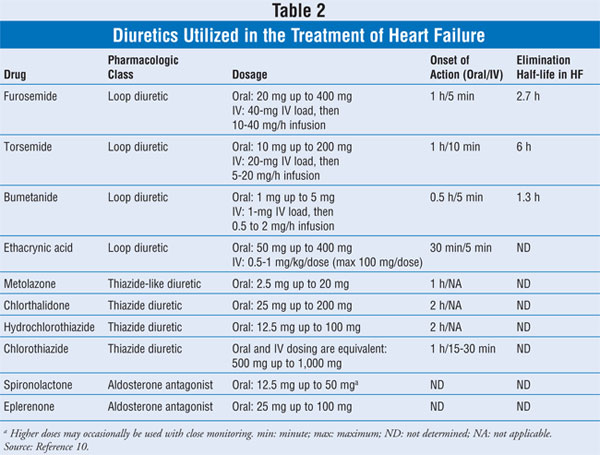
Loop Diuretics:
Loop diuretics remain the mainstay therapy for fluid removal in patients with
HF. They exert their action by inhibiting the sodium-potassium-chloride
cotransport system located within the thick ascending limb of the loop of
Henle. Patients with chronic HF tend to have a decreased response to normal
doses of loop diuretics secondary to reduced renal blood flow, increased
sodium reabsorption at sites downstream (distal tubule and collecting duct)
due to the activation of RAAS and the sympathetic nervous system, and possible
delayed intestinal absorption.11
The four loop diuretics used
in the treatment of HF are furosemide, bumetanide, torsemide, and ethacrynic
acid (TABLE 2). Ethacrynic acid is rarely used, but is reserved for
patients with true type-1 hypersensitivity reaction to sulfa drugs. There are
minimal clinically significant differences between furosemide, bumetanide, and
torsemide.11 One issue with furosemide is that its bioavailability
is 11% to 90% with erratic oral absorption; thus, patients who do not respond
to oral furosemide might benefit from switching to bumetanide or torsemide.
10 Murray et al illustrated with an open-label, randomized trial in 234
patients with chronic HF that patients who are treated with torsemide have
less fatigue and lower rate of readmission by 52% for HF at one year compared
to those treated with furosemide.12
Intravenous diuretics are more
potent than their equivalent oral doses, especially with severe or
decompensated HF due to significant interstitial edema of the gastrointestinal
tract. It is suggested that continuous infusion of loop diuretics has
potential advantages over intermittent bolus diuretic administration in
patients with severe HF, such as preventing postdiuretic sodium-chloride
retention, in addition to being more efficient and having less potential for
toxicity.13,14
The most common adverse
effects associated with loop diuretics include skin rashes, interstitial
nephritis, ototoxicity, gout, metabolic alkalosis, volume depletion,
hypokalemia, hypomagnesemia, hyperuricemia, and azotemia.10
Thiazide Diuretics:
Thiazide diuretics inhibit sodium reabsorption by blocking the electroneutral
sodium-chloride cotransporter at the distal tubule. Because the cotransporter
only reabsorbs about 5% of filtered sodium, thiazide diuretics are less
efficacious than loop diuretics in producing diuresis or natriuresis. The four
thiazide diuretics utilized in the U.S. (chlorthalidone, chlorothiazide,
hydrochlorothiazide, and metolazone) are described in TABLE 2. These
agents possess very similar characteristics except for chlorothiazide, which
may be administered IV.10
Thiazide diuretics are usually
added to a loop diuretic regimen to augment the diuresis in patients with
refractory edema. This combination appears to be synergistic by possible
multiple mechanisms including decreased postdiuretic sodium chloride
retention, decreased sodium transport along the proximal tubule through
inhibition of carbonic anhydrase, and by inhibition of sodium reabsorption at
the distal convoluted tubule.15,16 The effective doses of thiazide
diuretics vary among patients, with some patients requiring daily
administration and others only requiring them once weekly. Another approach is
to use the combination for a short, fixed period of time and then revert
to monotherapy with a loop diuretic when the patient achieves euvolemia.
The most common adverse
effects associated with the thiazide diuretics include skin rashes,
interstitial nephritis, pancreatitis, gout, alkalosis, volume depletion,
hypokalemia, hypomagnesemia, hypercholesterolemia, hypertriglyceridemia,
hyperglycemia in diabetics, and azotemia.10
Aldosterone Antagonists:
Although aldosterone antagonists are not technically classified as diuretics,
their adjunctive use in HF relates to their additive diuretic effect in
combination with other drugs, decreasing the cardiac workload and reducing
edema. Patients with HF have increased RAAS activity, and the direct local
effect of aldosterone on the heart may include hypertrophy, fibrosis, and
proarrhythmia.17,18 There are currently two aldosterone antagonists
available in the U.S.--spironolactone and eplerenone (TABLE 2).
The Randomized Aldactone
Evaluation Study (RALES) evaluated spironolactone, which competes with
aldosterone for the mineralocorticoid receptor sites in the distal renal
tubules, increasing sodium chloride and water excretion while conserving
potassium and hydrogen ions. The clinical trial showed that the addition of
spironolactone to patients with systolic dysfunction and NYHA class III
resulted in a 30% reduction in mortality and a 35% reduction in
hospitalization for HF; nevertheless, patients treated with spironolactone had
a significant reduction in systolic and diastolic blood pressure and a lower
incidence of hypokalemia, but a dose-related increased risk of hyperkalemia.
19 However, due to nonselective binding of spironolactone to the
androgen and progesterone receptors, endocrine adverse effects such as
gynecomastia, breast pain, menstrual irregularities, impotence, and decreased
libido were experienced.
Eplerenone, a more selective
agent to the mineralocorticoid receptor, has fewer endocrine adverse effects
than spironolactone. Eplerenone was evaluated in the Eplerenone Post-Acute
Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS),
which enrolled patients who had myocardial infarction and reduced LV systolic
function (LV ejection fraction [LVEF] ?40%). Patients who were treated
with eplerenone had significant reduction in all causes of mortality, which
was entirely due to the reduction in cardiovascular mortality.20
The use of an aldosterone
antagonist requires very careful monitoring for renal function and potassium,
as the incidence of fetal hyperkalemia has increased since the RALES
publication.21 Special consideration should be taken in patients
with other comorbidities such as significant renal dysfunction, advanced age,
and diabetes.
Investigational Agents
Vasopressin
Antagonists: The decreased activation of mechanoreceptors at the LV,
carotid sinus, aortic arch, and renal afferent arterioles by the decreased
cardiac output in patients with HF will lead to enhanced release of arginine
vasopressin and stimulation of thirst.3 The three vasopressin
receptors are V1a, V1b, and V2. V2
receptors are located at the basolateral membrane of the distal nephron and
renal collecting duct.22 Selectively blocking the V2
receptors in patients with HF improves urine output and increases serum sodium
concentration,23 while V1a and V1b receptors
principally cause vasoconstriction and mediate adrenocorticotropin release,
respectively.24
A recent multicenter,
randomized, double-blind study evaluated oral tolvaptan, a selective V2
receptor antagonist, in patients with HF and systolic dysfunction. In this
study there was no difference between tolvaptan and placebo on the LV volume,
but there was a reduction in the combined end point of mortality and HF in the
patients who were treated with tolvaptan.25
Conivaptan, an agent that
blocks both V1a and V2, is currently FDA approved for
the treatment of hyponatremia. It was evaluated in a small study in patients
with advanced HF. A single IV dose of conivaptan resulted in significant
reduction in the preload and an increase in urine output compared to placebo.
26
Large trials are needed to
evaluate vasopressin antagonists in HF, which have a potential benefit in
inducing aquaresis without an increase in the activation of RAAS.27
Adenosine Antagonists:
Adenosine reduces renal blood flow and decreases the glomerular
filtration rate (GFR) in patients with HF.28 Four adenosine
receptors (ARs) have been identified: A1, A2A, A2B
, and A3.29 According to Elkayam et al, direct infusion
of adenosine into the renal arteries of patients with chronic HF leads to
reduction of renal blood flow.30 This mechanism suggests that
blocking the ARs in the kidneys may enhance the renal blood flow in patients
with chronic HF. Gottlieb et al showed in small group of patients with HF that
administering an A1 antagonist resulted in increased sodium
excretion without a decrease in GFR compared to furosemide.31
Givertz et al evaluated the effects of the A1 antagonist KW-3902 in
patients with acute decompensated HF and renal impairment or diuretic
resistance.32 The infusion of KW-3902 resulted in increased urine
output and decreased serum creatinine compared to placebo.
These few studies showed
promising results, but safety needs to be established in humans because ARs
are found in all organs and play a key role in regulating metabolism. Larger
studies are under way (PROTECT-1 and PROTECT-2) to evaluate the potential use
of this new class of medication in patients with HF.
Conclusion
Treatment of HF has
changed dramatically during the past 20 years, but diuretic agents remain an
essential component in removal of excess fluids. While loop diuretics are
regarded as first-line therapy, the addition of thiazide diuretics may be
required in patients with advanced HF. Patients with systolic LV dysfunction
with NYHA class III and IV will also benefit from addition of aldosterone
antagonists; however, a close monitoring of renal function and serum potassium
level is required. Novel agents such as vasopressin antagonists and adenosine
antagonists are currently being evaluated, with promising results. When
treating HF with diuretics, care must be taken to not unload too much volume,
which may result in cardiac output depression.
REFERENCES
1. American Heart
Association (AHA). Heart Disease and Stroke Statistics: 2008 Update.
Dallas, TX: AHA; 2008.
www.americanheart.org/downloadable/heart/1200078608862HS_Stats%202008.final.pdf.
Accessed May 8, 2008.
2. NHLBI. Heart
failure. Who is at risk for heart failure?
www.nhlbi.nih.gov/health/dci/Diseases/Hf/HF_WhoIsAtRisk.html. Accessed May 8,
2008.
3. Schrier RW, Abraham
WT. Hormones and hemodynamics in heart failure. N Engl J Med.
1999;341:577-585.
4. Vogl A. The
discovery of the organic mercurial diuretics. Am Heart J.
1950;39:881-883.
5. Lloyd-Jones DM,
Larson MG, Leip EP, et al. Lifetime risk for developing congestive heart
failure: the Framingham Heart Study. Circulation. 2002;106:3068-3072.
6. Gheorghiade M, Bonow
RO. Chronic heart failure in the United States: a manifestation of coronary
artery disease. Circulation. 1998;97:282-289.
7. Velazquez EJ,
Pfeffer MA. Acute heart failure complicating acute coronary syndromes: a
deadly intersection. Circulation. 2004;109:440-442.
8. Nohria A, Mielniczuk
LM, Stevenson LW. Evaluation and monitoring of patients with acute heart
failure syndromes. Am J Cardiol. 2005;96:32G-40G.
9. ACC/AHA 2005
guideline update for the diagnosis and management of chronic heart failure in
the adult--summary article. Circulation. 2005;112:1825-1852.
10. Brater DC. Diuretic
therapy. N Engl J Med. 1998;339:387-394.
11. Brater DC, Day B,
Burdette A, et al. Bumetanide and furosemide in heart failure. Kidney Int.
1984;26:183-189.
12. Murray MD, Deer MM,
Ferguson JA, et al. Open-label randomized trial of torsemide compared with
furosemide therapy for patients with heart failure. Am J Med.
2001;111:513-520.
13. Salvador DR, Rey
NR, Ramos GC, Punzalan FE. Continuous infusion versus bolus injection of loop
diuretics in congestive heart failure. Cochrane Database Syst Rev.
2004;(1):CD003178.
14. Ross A, Hershberger
RE, Ellison DH. Diuretics in congestive heart failure. In: Feldman HM, ed.
Heart Failure: Pharmacologic Management. Malden, MA: Blackwell Futura;
2006:1-16.
15. Ellison DH. The
physiologic basis of diuretics synergism: its role in treating diuretic
resistance. Ann Intern Med.1991;114:886-894.
16. Oster JR, Epstein
M, Smoller S. Combined therapy with thiazide-type and loop diuretics agents
for resistant sodium retention. Ann Intern Med. 1983;99:405-406.
17. Lijnen P, Petrov V.
Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol.
2000;32:865-879.
18. Fullerton MJ,
Funder JW. Aldosterone and cardiac fibrosis: in vitro studies. Cardiovasc
Res. 1994;28:1863-1867.
19. Pitt B, Zannad F,
Remme WJ, et al. The effect of spironolactone on morbidity and mortality in
patients with severe heart failure. Randomized Aldactone Evaluation Study
Investigators. N Engl J Med.1999;34:709-717.
20. Salvador DR, Rey
NR, Ramos GC, Punzalan FE. Continuous infusion versus bolus injection of loop
diuretics in congestive heart failure. Cochrane Database Syst Rev.
2004;(1):CD003178.
21. Juurlink D, Mamdani
M, Lee D, et al. Rates of hyperkalemia after publication of Randomized
Aldactone Evaluation Study. N Engl J Med. 2004;351:543-551.
22. Carmichael MC,
Kumar R. Molecular biology of vasopressin receptors. Semin Nephrol.
1994;14:341-348.
23. Abraham WT, Oren
RM, Crisman TS, et al. Effect of an oral, nonpeptide, selective V2 receptor
vasopressin antagonist in patients with chronic heart failure. J Am Coll
Cardiol. 1997;29(suppl A):169A.
24. Greenberg A,
Verbalis JG. Vasopressin receptor antagonists. Kidney Int.
2006;69:2124-2130.
25. Udelson JE, McGrew
FA, Flores E, et al. Multicenter, randomized, double-blind, placebo-controlled
study on the effect of oral tolvaptan on left ventricular dilation and
function in patients with heart failure and systolic dysfunction. J Am Coll
Cardiol. 2007;49:2151-2159.
26. Udelson JE, Smith
WB, Hendrix GH, et al. Acute hemodynamic effects of conivaptan, a dual V(1A)
and V(2) vasopressin receptor antagonist, in patients with advanced heart
failure. Circulation. 2001;104:2417-2423.
27. Hirano T, Yamamura
Y, Nakamura S, et al. Effects of the V2 receptor antagonist OPC-41061 and the
loop diuretic furosemide alone and in combination in rats. J Pharmocol Exp
Ther. 2000;292:288-294.
28. Dzau VJ. Renal and
circulatory mechanisms in congestive heart failure. Kidney Int.
1987;31:1402-1415.
29. Fredholm BB,
IJzerman AP, Jacobson KA, et al. International Union of Pharmacology. XXV.
Nomenclature and classification of adenosine receptors. Pharmacol Rev.
2001;53:527-552.
30. Elkayam U, Mehra A,
Cohen G, et al. Renal circulatory effects of adenosine in patients with
chronic heart failure. J Am Coll Cardiol. 1998;32:211-215.
31. Gottlieb SS,
Skettino SL, Wolff A, et al. Effect of BG9719 (CVT-124), an A1-receptor
adenosine antagonist, and furosemide on GFR and natriuresis in patients with
congestive heart failure. J Am Coll Cardiol. 2000;35:56-59.
32. Givertz MM, Massie
BM, Fields TK, et al. The effects of KW-3902, an adenosine A1-receptor
antagonist, on diuresis and renal function in patients with acute
decompensated heart failure and renal impairment or diuretic resistance. J
Am Coll Cardiol. 2007;50:1551-1560.
To comment on this article, contact
rdavidson@jobson.com.


