US Pharm.
2007;32(7):48-54.
The aggressive medical management strategies
for chronic disease states, such as hypertension, cardiovascular disease, and
diabetes, are a characteristic component of Western medicine. Scientific data
demonstrate that medicalization improves symptom-related quality of life and
may reduce the risk of death. In April 2007, the Medical Expenditure Panel
Survey reported that the number of purchased prescriptions had increased by
one billion in seven years.1 Despite the progress made in drug
development and pharmacotherapeutic management of chronic disease, there has
been a trend toward increased drug-related adverse effects, adverse drug
reactions, and drug-related toxicity. Health care professionals, particularly
pharmacists, have a special responsibility to monitor for drug-induced
toxicities to ensure optimal safety and efficacy with aggressive medication
management strategies. This article explores the relationship between
pharmacotherapy and pulmonary adverse effects from frequently prescribed
agents used to treat common chronic diseases.
Beta Adrenoceptor
Antagonists
Beta adrenoceptor
antagonists, or beta-blockers, inhibit sympathetic stimulation by
competitively antagonizing catecholamines at beta-1 and beta-2 adrenergic
receptors found in the cardiac myocytes and vasculature. Cardioselective
agents have greater affinity for beta-1 receptors, which are found primarily
in the heart, while nonselective agents block beta-2 receptors, which are
predominantly found in blood vessels and bronchial smooth muscle.
Beta-blockers have been used
in the management of several chronic disease states, including glaucoma,
ocular hypertension, myocardial infarction, congestive heart failure, and
hypertension.2-4 Generally, this class of drugs is well tolerated
when administered orally, topically, or intravenously. However, concerns
regarding pulmonary adverse effects, particularly beta-blocker-associated
bronchospasm, have triggered some diffidence in prescribing beta-blockers.
These pulmonary adverse effects can be seen regardless of the route of
administration or the presence of pulmonary diseases (Table 1).
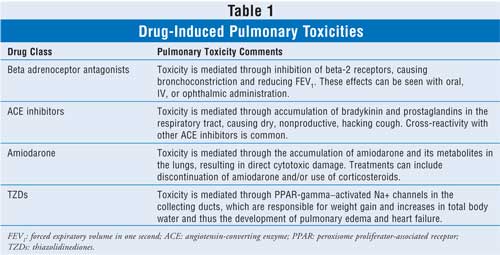
Pulmonary adverse effects have
been reported with oral and intravenous administration of beta-blockers. A
meta-analysis of 19 studies evaluated the effects of either single-dose
treatments or therapy of longer duration (two days to 3.3 months) with the
cardioselective beta-blockers atenolol, metoprolol, and bisoprolol in patients
with chronic obstructive pulmonary disease (COPD). The baseline forced
expiratory volume in one second (FEV1) was 2.4 ± 0.15 L
in the treatment group and 2.42 ± 0.2 L in the placebo group. The analysis
combined all agents into one group and showed a 7.46% reduction in FEV1
(CI 5.59-9.32%). Moreover, the FEV1 was only increased by 4.63%
after administration of the beta-agonist. The number of patients with
respiratory symptoms did not significantly increase (CI -0.02-0.03%).5,6
A retrospective cohort study
evaluated the tolerability of the nonselective beta-blocker carvedilol in 89
patients with COPD who were taking the medication for at least three months.
Carvedilol was tolerated in 85% of patients; however, the authors of this
trial did not address reversible airflow obstruction or reasons why patients
did not tolerate the drug.7
Ophthalmic administration of
beta-blockers can also cause pulmonary effects. After topical administration,
beta-blockers drain into the lacrimal ducts, where systemic absorption occurs
via the nasal mucosa and the facial and ophthalmic veins. Unlike the oral
route, which undergoes extensive first-pass hepatic metabolism, the topical
route bypasses this metabolism, allowing higher concentrations to reach
systemic circulation.8 Furthermore, the extent and timing of
beta-adrenoceptor occupancy have an important role in determining the systemic
effects of these drugs.9 A recent study evaluated the effects of
long-term treatment with topical timolol on bronchial reactivity in patients
with glaucoma who did not have pulmonary disease. The mean FEV1 at
three-year and four-year follow-up was decreased since baseline in the
timolol-treated group compared with the laser-treated group (P <.05 and
P = .052, respectively). These effects were not reversed upon
discontinuation of timolol.10
Although beta-blockers have
been shown to have undesirable pulmonary effects, anecdotally, they have been
shown to decrease morbidity and mortality in certain indications, such as use
in patients following myocardial infarction and congestive heart failure. The
relevance of a decreased FEV1 and whether this truly leads to
frank, symptomatic bronchospasm at commonly used doses needs to be clinically
validated.
Angiotensin-Converting
Enzyme (ACE) Inhibitors
ACE inhibitors are
a frequent cause of cough, with a reported incidence ranging from 1% to 12%.
11-14 This variation in frequency depends on the specific ACE inhibitor
used and the patient population. For instance, captopril-associated cough has
a reported incidence of 2%, whereas the incidence of perindopril- and
ramipril-associated cough can be as high as 12%.15,16 The patient
populations that are most susceptible to ACE inhibitor-associated cough are
the elderly (older than 60), women, and certain ethnicities, including
Chinese, Japanese, Indian, and black.12,16-21
ACE inhibitor-induced cough
often begins as a throat tickle and progresses to a dry, nonproductive,
hacking cough that is not associated with any other pulmonary abnormality
(i.e., chest x-ray, spirometry).15 Symptoms may begin within one
day or as late as 10 months after initiation of the ACE inhibitor.20
The exact mechanism is not fully elucidated, but most theories attribute
cough to the accumulation of bradykinin and prostaglandins in the respiratory
tract.22-25 ACE inhibitors prevent ACE from degradating bradykinin,
resulting in bradykinin accumulation which, according to observations from
experimental studies, results in cough.26-28 With continued ACE
inhibitor therapy, the cough may resolve within a few weeks, but more often,
discontinuation of the ACE inhibitor and initiation of an alternative therapy
is required. In the latter case, symptoms resolve within a few days of
discontinuation; however, some reports describe as long as one month for
symptom resolution.16,29,30 Additionally, cross-reactivity with
other ACE inhibitors is common and should be avoided.16,17,19,29,31
Angiotensin II receptor
blockers may be considered an alternative therapy for patients who are
intolerant of ACE inhibitors (due to cough), since bradykinin degradation is
not inhibited. Adjunctive therapies to treat ACE inhibitor-induced cough, such
as elemental iron, theophylline, cromolyn sodium, sulindac, and bupivacaine,
have been investigated; however, these therapies have proven to be ineffective
and counterproductive to the underlying disease state (e.g., sulindac and
hypertension) or have not been recommended for long-term use.16,17,32-34
Amiodarone
Amiodarone is a
class III antiarrhythmic agent commonly used to manage atrial fibrillation,
supraventricular tachyarrhythmias, ventricular fibrillation, and ventricular
tachycardia. Amiodarone has an extensive adverse drug-effect profile involving
the liver, thyroid, cornea, skin, and neuromuscular system, thereby limiting
its use.35
Amiodarone pulmonary toxicity (APT)
is a common lung injury, with a prevalence of between 1% and 15% of the
treated population.36 An estimated incidence rate of one in 1,000
to 2,000 patients treated per year has been reported.35,37 Symptoms
of APT include progressive dyspnea, malaise, nonproductive cough, and
pleuritic chest pain. Case reports of APT were first published in the early
1980s, and since then, literature suggests that APT is a current problem in
clinical practice.36,38-40
APT is most commonly seen in
men older than 40. Risk increases with age, preexisting lung disease, and
dosage and duration of therapy.41 Some speculate that risk of APT
only develops with higher daily doses of amiodarone (e.g., >500 mg/day);
however, the literature has shown that there is an elevated risk of APT in
patients who take up to 200 mg/day.42 On average, APT develops in
10% of patients taking more than 500 mg/day; the risk of APT is reduced to
0.5% in patients taking up to 200 mg/day.38,43,44
The proposed mechanism of APT is
driven by the accumulation of amiodarone and its metabolite,
desethylamiodarone, in the tissues of the lung. This accumulation leads to
direct cytotoxic damage and indirect immunologic reactions.35,45
APT can be identified through chest radiography (chest x-ray), CT scan, and
pulmonary function tests.36
Most cases of APT develop
within the first 1.5 years of treatment.36 Time to onset can be
quicker in patients taking greater doses. Some case reports detail patients
developing APT eight months after the discontinuation of amiodarone; this
delayed adverse effect may be related to the accumulation of amiodarone in the
lung.36,46
Treatment of APT includes
discontinuation of amiodarone, and clinical improvement typically requires one
to two months. Improvement in radiologic findings and pulmonary function tests
typically lag behind clinical improvements and may take up to 18 months to
resolve.47,48
Controversy exists over the
addition of corticosteroids for the management of APT after discontinuation of
amiodarone. The endorsement for the use of corticosteroids in managing APT has
been determined by case reports and expert opinion, and there is some guidance
available for their use: (1) initial dosage should range from 0.75 to 1 mg/kg
of prednisolone or equivalent; (2) initial dosage should be maintained until
definite clinical and radiographic response is obtained; otherwise, increased
doses should be considered; (3) corticosteroid taper should be slow; (4)
estimated duration could range from six to 12 months; (5) patients should be
monitored after discontinuation of corticosteroids for possible recurrence of
APT.36,48-50
Thiazolidinediones (TZDs)
TZD-induced
pulmonary edema and heart failure are of growing concern, since the incidence
and prevalence of type 2 diabetes continues to increase worldwide and TZDs
gain acceptance as guideline-recommended first-line therapy following diet and
lifestyle changes in patients with diabetes.51 Landmark, randomized
trials are also reporting an increase in TZD-induced edema and heart failure
in patients with and without macrovascular disease primarily from expanded
intracellular volume due to altered handling of sodium in the renal collecting
duct.52-54
Edema has been reported in 7.2% to
12.4% of patients receiving a TZD in combination with other oral hypoglycemics
and in up to 15.2% of those using a TZD with insulin.55,56 The
increased frequency of heart failure in patients taking both TZDs and insulin
has led both European and Canadian health agencies to consider insulin therapy
as a contraindication to TZDs.57,58
Heart failure has been reported in
9.9% of patients with uncontrolled type 2 diabetes and New York Heart
Association (NYHA) class II or III heart failure using pioglitazone plus
insulin. In addition, heart failure has been reported in 6% of patients with
uncontrolled type 2 diabetes and NYHA class I or II heart failure receiving
rosiglitazone in addition to hypoglycemic medications.55,56
The time of onset to edema and
heart failure with TZDs ranges from several days to weeks and remits with
discontinuation and aggressive diuresis with furosemide. This strongly
suggests that TZDs are responsible for new edema, the exacerbation of existing
heart failure, and new cases of heart failure. Risk factors for TZD-induced
edema or heart failure include established heart failure, left ventricular
hypertrophy, valvular disease, chronic renal failure or insufficiency, age 70
or older, atherosclerosis, hypertension, obesity, history of diabetes 10 years
or longer, and concurrent insulin or sulfonylurea use.59-62 The
American Diabetes Association allows patients with stage I or II heart failure
to receive TZDs;51 however, other experts are prudent and caution
that TZDs should be avoided in all heart failure patients, including those in
stage I or II.62
A reasonable pharmacologic
explanation and recent finding suggests that peroxisome proliferator-activated
receptor-gamma (PPAR-gamma)–activated Na+ channels in the collecting ducts are
responsible for weight gain and increased total body water with TZDs.
PPAR-gamma agonists promote enhanced sodium absorption in the collecting duct,
which results in fluid retention and plasma volume expansion, which in turn
can result in new heart failure. This finding also explains the trend toward
small reductions in hemoglobin and hematocrit, which can result in a mild
dilutional anemia that has been associated with TZDs. Selective deletion of
the PPAR-gamma–activated channel through genetic deletion in mice fails to
result in increases in total body water in the presence of a TZD. Amiloride, a
selective inhibitor of sodium absorption in the collecting duct, also prevents
increases in plasma volume and total body water in the presence of a
PPAR-gamma agonist.63 Other mechanisms include reversible
PPAR-gamma–mediated pulmonary endothelial hyperpermeability, which is four
times greater than normal at doses in the higher therapeutic range, with the
clinical implication of predisposing patients to pulmonary edema.42
This combination of PPAR-gamma agonist–induced increases in sodium
reabsorption in the collecting duct and plasma volume expansion along with
enhanced pulmonary endothelial hyperpermeability may result in clinically
significant pulmonary edema and heart failure, especially in patients with a
history of heart failure, established cardiac disease, or renal failure.
Taken together, TZDs increase
the risk of heart failure across the entire spectrum of high- and low-risk
patients with diabetes, including those with established macrovascular
disease, those without macrovascular disease, and even those with impaired
fasting glucose or impaired glucose tolerance.
Conclusions
The improvements in
chronic disease outcomes that stem from aggressive medical management require
increased pharmacist awareness and monitoring for the possibility of new
drug-induced adverse events--in this case, a review of the mechanisms
associated with drug-induced pulmonary adverse events. Beta-blockers, ACE
inhibitors, amiodarone, and TZDs account for the majority of drug-related
pulmonary toxicity but are just some of the agents known to cause or
exacerbate pulmonary adverse events. Drug-associated pulmonary adverse events
occur more frequently in patients with predictable underlying risk factors,
such as established pulmonary disease, heart failure, or advanced age, and can
present in many ways, such as dyspnea on exertion, pleuritic chest pain, or
dry cough. In order to ensure optimal medication management, pharmacists
should take an active role in identifying patients at risk for drug-induced
pulmonary toxicities, monitoring patients, and providing patient education.
Pharmacists may also help patients make informed decisions regarding the
long-term use of medications with established pulmonary toxicity profiles.
Some patients may not be able to readily interpret and apply concepts of
drug-induced toxicity, so pharmacist-driven coaching and education can be
helpful when patients are deliberating the trade-offs of symptom-related
mortality or drug-related morbidity.
References
1. Stagnitti, M. N.
Trends in Outpatient Prescription Drug Utilization and Expenditures: 1997 and
2004. Statistical Brief #168. April 2007. Agency for Healthcare Research and
Quality. Rockville, Md. Available at:
www.meps.ahrq.gov/mepsweb/data_files/publications/st168/stat168.pdf.
2. Zimmerman TJ.
Topical ophthalmic beta blockers: a comparative review. J Ocular Pharmacol
. 1993;9(4):373-384.
3. Freemantle N,
Cleland J, Young P, et al. beta Blockade after myocardial infarction:
systematic review and meta regression analysis. BMJ. 1999;318:1730-1737.
4. Doughty RN, Rodgers
A, Sharpe N, MacMahon S. Effects of beta-blocker therapy on mortality in
patients with heart failure. A systematic overview of randomized controlled
trials. Eur Heart J. 1997;18:560-565.
5. Salpeter SR,
Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with
reactive airway disease: a meta-analysis. Ann Intern Med.
2002;137:715-725.
6. Salpeter S, Ormiston
T, Salpeter E. Cardioselective beta-blockers for chronic obstructive pulmonary
disease. Cochrane Database Syst Rev. 2005(4):CD003566.
7. Krum H, Ninio D,
MacDonald P. Baseline predictors of tolerability to carvedilol in patients
with chronic heart failure. Heart. 2000;84:615-619.
8. Juzych MS, Zimmerman
TJ. b-Blockers. Philadelphia, PA: Lippincott-Raven; 1997.
9. Rait JL. Systemic
effects of topical ophthalmic beta-adrenoceptor antagonists. Aust N Z J
Ophthalmol. 1999;27:57-64.
10. Gandolfi SA, Chetta
A, Cimino L, et al. Bronchial reactivity in healthy individuals undergoing
long-term topical treatment with beta-blockers. Arch Ophthalmol.
2005;123:35-38.
11. Speirs C, Wagniart
F, Poggi L. Perindopril postmarketing surveillance: a 12 month study in 47,351
hypertensive patients. Br J Clin Pharmacol. 1998;46:63-70.
12. Kostis JB, Shelton
B, Gosselin G, et al. Adverse effects of enalapril in the Studies of Left
Ventricular Dysfunction (SOLVD). SOLVD Investigators. Am Heart J.
1996;131(2):350-325.
13. Lacourciere Y,
Lefebvre J, Nakhle G, et al. Association between cough and angiotensin
converting enzyme inhibitors versus angiotensin II antagonists: the design of
a prospective, controlled study. J Hypertens Suppl. 1994;12:S49-S53.
14. Fletcher AE, Palmer
AJ, Bulpitt CJ. Cough with angiotensin converting enzyme inhibitors: how much
of a problem? J Hypertens Suppl. 1994;12:S43-S47.
15. Stoller JK,
Elghazawi A, Mehta AC, Vidt DG. Captopril-induced cough. Chest.
1988;93:659-661.
16. Just PM. The
positive association of cough with angiotensin-converting enzyme inhibitors.
Pharmacotherapy. 1989;9:82-87.
17. Israili ZH, Hall
WD. Cough and angioneurotic edema associated with angiotensin-converting
enzyme inhibitor therapy. A review of the literature and pathophysiology.
Ann Intern Med. 1992;117:234-242.
18. Woo J, Chan TY. A
high incidence of cough associated with combination therapy of hypertension
with isradipine and lisinopril in Chinese subjects. Br J Clin Pract.
1991;45:178-180.
19. Chan P, Tomlinson
B, Huang TY, et al. Double-blind comparison of losartan, lisinopril, and
metolazone in elderly hypertensive patients with previous
angiotensin-converting enzyme inhibitor-induced cough. J Clin Pharmacol
. 1997;37:253-257.
20. Seedat YK, Randeree
IGH. Antihypertensive effect and tolerability of perindopril in Indian
hypertensive and type 2 diabetic patients. Clin Drug Invest.
1998;16:229-240.
21. Elliott WJ. Higher
incidence of discontinuation of angiotensin converting enzyme inhibitors due
to cough in black subjects. Clin Pharmacol Ther. 1996;60:582-588.
22. Burkart W. Severe
coughing during captopril and enalapril therapy. CMAJ. 1986;135:1070.
23. Sesoko S, Kaneko Y.
Cough associated with the use of captopril. Arch Intern Med.
1985;145:1524.
24. Semple PF, Herd GW.
Cough and wheeze caused by inhibitors of angiotensin-converting enzyme. N
Engl J Med. 1986;314:61.
25. Israel-Biet D,
Delaisements C, Chretien J. Enalapril-induced cough. Lancet. 1986;2:918.
26. Varonier HS,
Panzani R. The effect of inhalations of bradykinin on healthy and atopic
(asthmatic) children. Int Arch Allergy Appl Immunol. 1968;34:293-296.
27. Newball HH, Keiser
HR. Relative effects of bradykinin and histamine on the respiratory system of
man. J Appl Physiol. 1973;35:552-556.
28. Fuller RW, Dixon
CM, Cuss FM, Barnes PJ. Bradykinin-induced bronchoconstriction in humans. Mode
of action. Am Rev Respir Dis. 1987;135:176-180.
29. Inman WH.
Enalapril-induced cough. Lancet. 1986;2:1218.
30. Punzi HA. Safety
update: focus on cough. Am J Cardiol. 1993;72:45H-48H.
31. Rossetto BJ. Side
effect of captopril and enalapril. West J Med. 1987;146:102.
32. Lee SC, Park SW,
Kim DK, Lee SH, Hong KP. Iron supplementation inhibits cough associated with
ACE inhibitors. Hypertension. 2001;38:166-170.
33. Israili ZH, Hall
WD. Adverse effects of ACE inhibitors. Ann Intern Med.1993;118:315.
34. Aldis WL. Cromolyn
for cough due to angiotensin-converting enzyme inhibitor therapy. Preliminary
observations. Chest. 1991;100:1741-1742.
35. Raissy HH, Harkins
M, Marshik PL. Drug-Induced Pulmonary Diseases. In: Dipiro JT, ed.
Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York, NY:
McGraw-Hill; 2005.
36. Camus P, Martin WJ,
Rosenow EC. Amiodarone pulmonary toxicity. Clin Chest Med.
2004;25:65-75.
37. Rakita L, Sobol SM,
Mostow N, Vrobel T. Amiodarone pulmonary toxicity. Am Heart J.
1983;106(4 Pt 2):906-916.
38. Vorperian VR,
Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a
meta-analysis. J Am Coll Cardiol. 1997;30:791-798.
39. Singh SN, Fisher
SG, Deedwania PC, et al. Pulmonary effect of amiodarone in patients with heart
failure. The Congestive Heart Failure-Survival Trial of Antiarrhythmic Therapy
(CHF-STAT) Investigators (Veterans Affairs Cooperative Study No. 320). J Am
Coll Cardiol. 1997;30:514-517.
40. Rotmensch HH, Liron
M, Tupilski M, Laniado S. Possible association of pneumonitis with amiodarone
therapy. Am Heart J. 1980;100:412-413.
41. Kaushik S, Hussain
A, Clarke P, Lazar HL. Acute pulmonary toxicity after low-dose amiodarone
therapy. Ann Thorac Surg. 2001;72(5):1760-1761.
42. Idris I, Gray S,
Donnelly R. Rosiglitazone and pulmonary oedema: an acute dose-dependent effect
on human endothelial cell permeability. Diabetologia. 2003;46:288-290.
43. Ott MC, Khoor A,
Leventhal JP, et al. Pulmonary toxicity in patients receiving low-dose
amiodarone. Chest. 2003;123(2):646-651.
44. Morady F, Sauve MJ,
Malone P, et al. Long-term efficacy and toxicity of high-dose amiodarone
therapy for ventricular tachycardia or ventricular fibrillation. Am J
Cardiol. 1983;52:975-979.
45. Jessurun GA,
Boersma WG, Crijns HJ. Amiodarone-induced pulmonary toxicity. Predisposing
factors, clinical symptoms and treatment. Drug Saf. 1998;18:339-344.
46. Chendrasekhar A,
Barke RA, Druck P. Recurrent amiodarone pulmonary toxicity. South Med J
. 1996;89:85-86.
47. Olson LK, Forrest
JV, Friedman PJ, et al. Pneumonitis after amiodarone therapy. Radiology
. 1984;150:327-330.
48. Vernhet H, Bousquet
C, Durand G, Giron J, Senac JP. Reversible amiodarone-induced lung disease:
HRCT findings. Eur Radiol. 2001;11:1697-1703.
49. Sobol SM, Rakita L.
Pneumonitis and pulmonary fibrosis associated with amiodarone treatment: a
possible complication of a new antiarrhythmic drug. Circulation.
1982;65:819-824.
50. Bates N, Dawson J.
Suspected amiodarone-hypersensitivity pneumonitis. Can J Hosp Pharm.
1998;51:117-121.
51. Standards of
medical care in diabetes--2007. Diabetes Care. 2007;30(Suppl
1):S4-S41.
52. Dormandy JA,
Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events
in patients with type 2 diabetes in the PROactive Study (PROspective
pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled
trial. Lancet. 2005;366:1279-1289.
53. Gerstein HC, Yusuf
S, Bosch J, et al. Effect of rosiglitazone on the frequency of diabetes in
patients with impaired glucose tolerance or impaired fasting glucose: a
randomised controlled trial. Lancet. 2006;368:1096-1105.
54. Kahn SE, Haffner
SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or
glyburide monotherapy. N Engl J Med. 2006;355:2427-2443.
55. Product monograph.
Actos (pioglitazone hydrochloride) tablets. Deerfield, Ill: Takeda
Pharmaceuticals America, Inc.; 2007.
56. Product monograph.
Avandia (rosiglitazone maleate) tablets. Research Triangle Park, NC:
GlaxoSmithKline; 2007.
57. Yki-Jarvinen H.
Thiazolidinediones. N Engl J Med. 2004;351:1106-1118.
58. Cheng AYY, Fantus
IG. Thiazolidinedione-induced congestive heart failure. Ann Pharmacother
. 2004:38:817-820.
59. Nesto RW, Bell D,
Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart
failure: a consensus statement from the American Heart Association and
American Diabetes Association. Diabetes Care. 2004:27:256-263.
60. Niemeyer N, Janney
L. Thiazolidinedione-induced edema. Pharmacotherapy. 2002;22:924-929.
61. Kermani A, Garg A.
Thiazolidinedione-associated congestive heart failure and pulmonary edema.
Mayo Clin Proc. 2003;78:1088-1091.
62. Mudaliar S, Chang
A, Henry R. Thiazolidinediones, peripheral edema, and type 2 diabetes:
incidence, pathophysiology, and clinical implications. Endocr Pract.
2003;9:406-16.
63. Guan Y, Hao C, Cha
DR, et al. Thiazolidinediones expand body fluid volume through PPARgamma
stimulation of ENaC-mediated renal salt absorption. Nat Med.
2005;11:861-866.
To comment on this article, contact
editor@uspharmacist.com.



