US Pharm. 2017;42(3)(Specialty&Oncology suppl):24-27.
ABSTRACT: Bleeding disorders may result from platelet defects, enhanced fibrinolytic activity, or clotting-factor deficits. Clotting-factor deficits are the main complication observed in patients with hemophilia. Hemophilia has an estimated frequency of 1 in 10,000 births, with approximately 400,000 individuals affected globally.1 Because hemophilia A and B are X-linked chromosome disorders caused by mutations in clotting factors VIII or IX genes, respectively, replacement of those factors in hemophilia patients is paramount. It is important for practitioners to be knowledgeable regarding advances in hemophilia management in order to assist in the process of drug selection.
The human body’s coagulation cascade is an intricate system designed to halt bleeding at a site of vascular injury. This is done through complex interactions with the endothelium, platelets, procoagulant proteins, anticoagulant proteins, and fibrinolytic proteins. Furthermore, this clotting system reflects a balance of coagulation and anticoagulation in a process known as hemostasis. Maintenance of hemostasis is critical as the severity of bleeding events ranges from mild to fatal.1 The three key mechanisms of hemostasis include vascular constriction, assembly of the platelet plug, and fibrin formation.2 Bleeding disorders may result from platelet defects, enhanced fibrinolytic activity, or clotting-factor deficits. Clotting-factor deficits are the mainstay of complications observed in patients with hemophilia and the target of pharmacologic therapy addressed in this review.3
Hemophilia is broadly defined as a congenital deficiency in a clotting protein and is classified according to the clotting factor that is deficient.4 Hemophilia has an estimated frequency of 1 in 10,000 births, with approximately 400,000 individuals affected globally.1,5 The incidence of hemophilia is approximately 1 in 5,000 male births with 80% to 85% of cases being attributed to hemophilia A and 15% to 20% of cases categorized as hemophilia B.6 Hemophilia A and B (caused by mutations in clotting factors VIII or IX genes, respectively) are X-linked recessive disorders that are caused by gene mutations located on the X chromosome. Males, who have only one X chromosome, only require one copy of the gene mutation to be present for the disease to exist. In females, who have two X chromosomes, the mutation must be present in both X chromosomes for them to be affected.4 Other bleeding irregularities such as von Willebrand disease, hemophilia C, and those associated with medication use are also well documented but are not the focus of this review.
Clinical Presentation and Diagnoses
Individuals with hemophilia may experience various signs of internal and/or external bleeding (TABLE 1). Hemophilia should be suspected in patients with a history of easy bruising/bleeding early in childhood, spontaneous bleeding, and excessive bleeding following trauma or surgery.1 Considering that hemophilia is a congenital disease, it is imperative that clinicians diagnose prenatally if hemophilia is suspected.4 A positive family history of bleeding may be helpful, but it is important to note that up to 50% of patients diagnosed with hemophilia have no significant family history of bleeding.

The severity of bleeding events generally correlates with the degree of factor VIII or IX deficiency, which guides treatment decisions such as choice of therapy and dosages necessary to achieve therapeutic serum-factor levels. Clotting- factor assays are required to diagnose bleeding disorders and will distinguish the type of hemophilia along with its severity.7 Factor levels are measured in units per milliliter and normal plasma levels are between 50% and 150%. Utilizing this reference range, patients can be further categorized into mild, moderate, or severe hemophilia based on factor levels (TABLE 2).1

History of Hemophilia Treatment
The treatment of hemophiliacs has evolved significantly over the past 50 years. Initially, the mainstay treatment modalities available for hemophilia were whole blood and fresh-frozen plasma.8 As a result of low factor VIII and IX levels in those products, most patients with severe hemophilia did not survive past early adulthood.9
The modern era of hemophilia management came into existence in the 1970s with the availability of plasma concentrates. Desmopressin, a synthetic medication that increases factor VIII and von Willebrand factor levels, provided a new, inexpensive method of safely treating patients with mild hemophilia A while minimizing risks of blood-borne infections.10 In the early 1980s, new challenges in the treatment of hemophilia arose as a vast number of patients with severe hemophilia became infected with HIV and hepatitis C transmitted by contaminated factor concentrates pooled from thousands of donors.10 As a result of this epidemic, the need for safer treatment of hemophilia developed. This lead to the cloning of factor VIII and IX genes, when the industrial production of recombinant factor VIII and factor IX became readily available. Additional measures have since been implemented to improve safety during the manufacturing process.10,11
Prophylaxis/Treatment of Hemophilia
Regular treatment to prevent anticipated bleeding in those with hemophilia is known as prophylaxis. The types of products that have historically been utilized vary (TABLE 3). This method of therapy has been shown to be useful; however, the length of time that one should remain on prophylaxis is unclear. There are many different protocols utilized to optimize clotting-factor levels at 1% or above; however, each method should be individualized as much as possible. Prophylactic therapy is commonly divided into several types. Continuous prophylaxis is administered over several months to years, while intermittent prophylaxis is given for shorter periods of time.12 Prophylactic therapy is key in the management of hemophilia and typically involves home medication administration two to three times per week. Considering the half-life of factors VIII (~12 hours) and IX (~18 hours), the addition of several longer acting coagulation–factor products promotes prophylactic therapy at home by requiring less frequent administration and potentially reducing cost as a limiting factor.
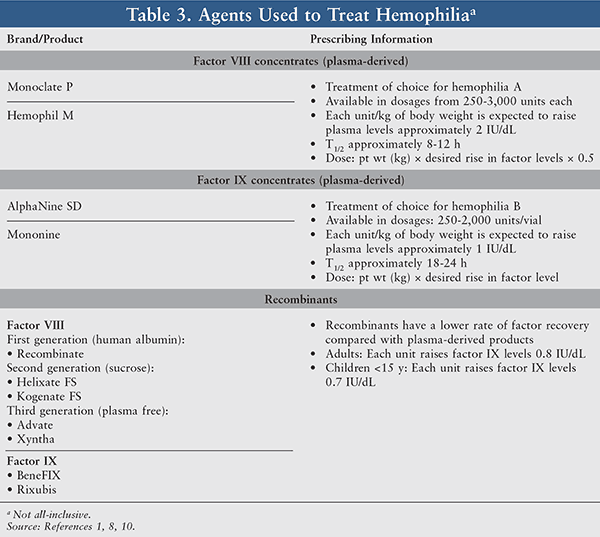
Those with acute bleeds should be treated as quickly as possible. Currently, the gold standard for on-demand treatment in hemophiliacs is immediate intravenous- factor replacement. This provides patients with rapid access to clotting factors resulting in decreased pain, dysfunction, long-term disability, and reduced readmissions due to hemophilia complications.1 Delays in treatment are associated with prolonged recovery and increased factor-concentrate usage, as well as permanent damage to joints, muscles, and other organs.1
Today, one of the most challenging complications of hemophilia therapy is the development of inhibitory antibodies against factors VIII and IX, which occurs in 25% to 30% of previously untreated patients with hemophilia A and 3% to 5% of patients with hemophilia B.10 The development of inhibitors renders replacement therapies ineffective, limits patient access to safe and effective care, and predisposes them to increased risk of morbidity and mortality. Bypassing agents, such as activated prothrombin complex concentrates (e.g., factor eight inhibitor bypassing activity) as well as recombinant activated factor VII (e.g., NovoSeven), have greatly improved the care of hemophilia patients with inhibitors. These agents do not restore normal pathways of the clotting cascade, but, instead, circumvent factors blocked by an inhibitor antibody to promote the body’s clot-forming process.
Trends/Future of Hemophilia Management
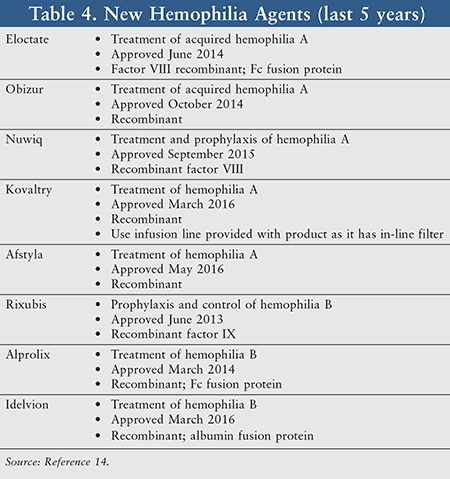
Recombinant factor products are produced via recombinant DNA technology, derived from hamster ovarian cells transfected with human factor VIII or IX genes. Clinical trials have demonstrated comparable efficacy between recombinant factor VIII products and plasma-derived products.13 As a result, recombinant products are frequently favored for the treatment of hemophilia, with several of the products being developed within the last few years (TABLE 4).14 As stated previously, these recombinant factors are being produced with longer half-lives, which may improve adherence. Most recently developed products require once- or twice-monthly injections.
The idea of gene therapy has been around for decades, although no gene-therapy product has come to market. The gene-therapy concept is that a patient can receive a one-time dose of a new clotting gene and not require a lifetime of multiple-factor injections. A potential downfall to this theory is that patients may reject the product because of the vector that it would be manufactured in. Within the past year, gene therapy has progressed, and it is closer to being an option due to clearance of gene-therapy testing in human patients.15
Pharmacist’s Role in Hemophilia Management
Pharmacists play an important role as a member of the treatment team in the management of patients with hemophilia. Those who have the expertise in this area are able to provide drug-utilization management, patient education, and services that promote medication adherence and improve outcomes.16 Their ability to provide dosage optimization, patient monitoring, and regimen revisions creates a unique niche for these pharmacists. Furthermore, they may assist providers with drug selection based upon availability/costs, presence of inhibitors, or severity of the bleeding event.
REFERENCES
1. Srivastava A, Brewer AK, Mauser-bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1-47.
2. Ho-tin-noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(suppl 1):56-65.
3. National Hemophilia Foundation. Medical and Scientific Advisory Council. MASAC recommendations concerning products licensed for the treatment of hemophilia and other bleeding disorders. New York, 2013 (revised November 2016). MASAC Document 246.
4. Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;3:15056.
5. Stonebraker JS, et al. A study of variations in reported haemophilia A prevalence around the world. Haemophilia. 2010;16(1):20-32.
6. Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998;59(4):288-294.
7. CDC. Hemophilia. www.cdc.gov/ncbddd/hemophilia/diagnosis.html. Accessed February 15, 2017.
8. McDaniel M. Treatment of hemophilia A and B. National Hemophilia Foundation, 2013. www.hemophilia.org. Accessed February 15, 2017.
9. Canadian Hemophilia Society. The history of hemophilia. www.hemophilia.ca/en/bleeding-disorders/hemophilia-a-and-b/the-history-of-hemophilia/. Accessed February 15, 2017.
10. Franchini M, Mannucci PM. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis. 2012;7:24.
11. Pipe SW. Recombinant clotting factors. Thromb Haemost. 2008;99(5):840-850.
12. About bleeding disorders. World Federation of Hemophilia. www.wfh.org/en/abd/prophylaxis/types-of-prophylaxis. Accessed February 15, 2017.
13. Lee C, Berntorp E, Hoots W, eds. Textbook of Hemophilia. 2nd ed. Chichester, West Sussex, UK: Wiley-Blackwell, 2010.
14. FDA. Approval of FDA-regulated products. www.fda.gov/newsevents/productsapprovals/default.htm. Accessed March 5, 2017.
15. FDA. FDA clears hemophilia A gene therapy, SB-525, for testing in patients. https://hemophilianewstoday.com/2017/01/06/sangamo-therapeutics-sb-525-to-treat-hemophelia-a-given-fda-investigational-new-drug-status/. Accessed March 5, 2017.
16. Blankenship CS. To manage costs of hemophilia, patients need more than clotting factor. Biotechnol Healthc. 2008;5(4):37-40.
To comment on this article, contact rdavidson@uspharmacist.com.



