US Pharm. 2020;45(7/8)( Specialty&Oncology suppl):3-6.
ABSTRACT: Hemophilia is one of the most common serious congenital clotting-factor deficiencies. In recent years, a number of new drugs have been FDA-approved to treat this bleeding disorder, and recent trial data on gene therapy are promising. In order to properly use new therapies, it is important for healthcare providers to understand the etiology of hemophilia, treatment pharmacology, and patient-specific factors. Hemophilia treatments are becoming more patient-specific compared with historical treatment using general human-sourced plasma. However, various obstacles must be overcome in order to select the optimal treatment for the individual patient. As drug experts, pharmacists will play an increasingly vital role in hemophilia treatment as targeted therapies increase in number and complexity; therefore, they must keep abreast of current research and developments in therapy.
Hemophilia, an X-linked genetic disorder, is one of the most common serious congenital clotting-factor deficiencies. Clotting factors, which circulate in the blood, are essential for the functioning of the coagulation cascade, a complex system designed to halt bleeding at a site of vascular injury. Because hemophilia is caused by mutations in clotting factors, replacement of the deficient factors in patients with this disorder is vital.1 In order to manage hemophilia safely, it is imperative that pharmacists have a strong foundation in the medications used to treat this disorder and actively participate in the drug-selection process. This article will focus on hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency).
Clinical Presentation and Diagnosis
Hemophilia patients experience both internal and external bleeding symptoms, such as bleeding, bruising, swelling, hemorrhage, and nerve compression.2 Hemophilia’s primary presentation is spontaneous bleeding—more specifically, interarticular bleeds. The degree of bleeding and the severity of the hemophilia diagnosis are dependent on the patient’s level of clotting-factor deficiency.
In order to properly coordinate care for hemophilia patients, early diagnostic measures are taken prior to the patient’s conception. If a female suspected to be a hemophilia carrier is pregnant or considering pregnancy, genetic counseling and testing are recommended to confirm or deny carrier status.3 If the mother declines genetic counseling or is confirmed to be a carrier, a diagnostic procedure is performed to determine the sex of the child. If the child is male or the sex is undetermined, measures will be taken at childbirth to avoid fetal scalp monitoring, blood sampling, and operative vaginal birth. These precautions commonly make cesarean section necessary.3 Once the child is born, confirmatory factor-level testing will be performed to establish a diagnosis.3
Patients with a factor level of 5 IU/dL to 40 IU/dL are classified as mildly factor deficient and will experience severe bleeds only with a major trauma or surgery. Patients with a level of 1 IU/dL to 5 IU/dL are classified as moderately factor deficient and will occasionally experience spontaneous bleeds, as well as extended bleeds during trauma or surgery. Severely factor-deficient patients (i.e., a level below 1 IU/dL) will experience chronic spontaneous joint and muscle bleeds (TABLE 1).2
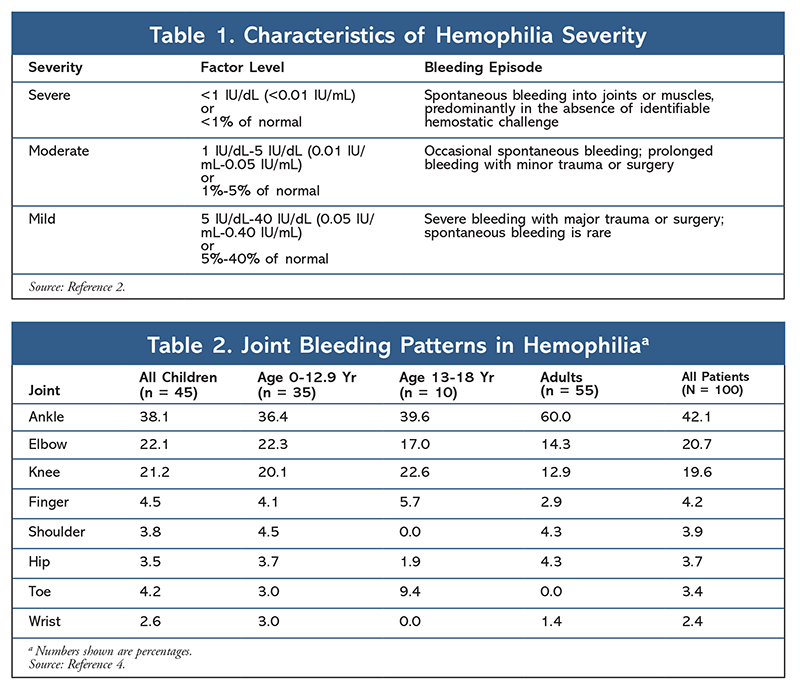
The joints most commonly affected by hemophilia are the ankle (38%), elbow (22%), and knee (21%) (TABLE 2).4 Continued intraarticular bleeding may lead to iron deposits in the synovial space, which can cause inflammation, synovial hypertrophy, and reduced range of motion.2,5 Patients often gradually discontinue activities that require use of these joints, resulting in a decreased quality of life.5
Prophylaxis and Treatment
Both moderate-hemophilia and severe-hemophilia patients are extremely likely to experience a type of joint damage known as hemophilic arthropathy.5 In order to prevent hemophilic arthropathy and other spontaneous bleeds, each week these patients receive prophylaxis consisting of multiple injections of concentrated amounts of the clotting factor they are deficient in. There are two types of hemophilia prophylaxis: primary prophylaxis, occurring before or at the start of the initial interarticular bleed, and secondary prophylaxis, administered once hemophilic arthropathy has progressed.5 Historically, hemophilia prophylaxis was given in standardized weight-based doses; all patients, regardless of variability in tendency to bleed, pharmacokinetics, and hemophilic arthropathy, received three or four doses of 20 U/dL to 40 U/dL every week.6 However, personalized prophylaxis has since become a standard of prophylactic treatment.7 Patients express satisfaction when their routine prophylactic treatments are adjusted in accordance with their activity level, bleed history, and lifestyle demands.7
Despite general success in the vast majority of patients with hemophilia, prophylaxis with conventional factor concentrates still faces some significant obstacles, including a short half-life.5 Factor concentrates with a short half-life lead to more frequent IV administration, reduced patient adherence, and increased treatment cost.8 In the past 3 years, novel technologies have led to increases in the half-life of factor concentrates and the development of six new FDA-approved hemophilia drugs (TABLE 3).9

Novel technologies include pegylation, fusion-protein technology, and single-chain technology. Pegylation increases the half-life 1.5-fold over standard half-life concentrates by reducing the binding of pegylated proteins to clearance proteins.8 Jivi, a pegylated recombinant antihemophilic factor, was FDA approved in 2018 for prophylaxis, treatment, and perioperative bleed management in previously treated hemophilia A patients.9 Fusion-protein technology extends the half-life of recombinant clotting factor 1.5-fold by fusing the concentrate with proteins that have a much longer half-life, such as albumin.8 The first FDA-approved medication for the treatment of hemophilia B that used this technology was Idelvion.9 As noted, another method used to combat the short half-life of hemophilia drugs is single-chain technology. This method also increases half-life up to 1.4 times longer than standard factor half-lives by increasing factor VIII affinity to von Willebrand factors.8
Another significant obstacle that hemophilia patients face is the development of inhibitors.5 Inhibitors, which are immunoglobulin G antibodies that specifically target factor VIII, can be completely inactivating or incompletely inactivating. Completely inactivating inhibitors commonly develop in patients with severe hemophilia A and lead to more frequent and serious bleeding complications.5 Hemlibra, a factor IXa– and factor X–directed antibody agent, is FDA approved for prophylaxis of hemophilia A patients who have inhibitors.9
Rebinyn has been FDA approved for the treatment of hemophilia B, and Sevenfact was FDA approved in early 2020 for the treatment and control of hemophilia A and hemophilia B.9 With the exception of Hemlibra, all of the agents summarized in TABLE 3 must be administered IV in a healthcare facility. Hemlibra may be administered subcutaneously either by a healthcare provider or by the patient who has received proper education and injection training.10
Looking Forward: Gene Therapy
The novel concept of gene therapy for hemophilia has been studied for decades. The reasoning is that one-time IV administration of a functional factor VIII would replace a patient’s dysfunctional factor VIII. This would lead to increased factor VIII production, fewer bleeding events, and, ultimately, a cure for hemophilia A.11
In May 2020, BioMarin Pharmaceuticals released updates on its phase 1/2 study of valoctocogene roxaparvovec gene therapy for severe hemophilia A, which began in 2015. This trial is investigating the long-term therapeutic effects of one-time administration of valoctocogene roxaparvovec with the intent to increase factor VIII production and reduce annualized bleed rates. There are two study cohorts: a large-dosage (6e13 vg/kg) group and a smaller-dosage (4e13 vg/kg) group. Trial updates for year 4 and year 3 of the large-dosage group and the smaller-dosage group, respectively, have been released. Both groups have successfully discontinued factor VIII prophylaxis after receipt of the one-time dose. Annualized bleed rates for patients in both groups are lower than their baseline pretreatment levels. These data are promising, and the FDA is reviewing the biologics license for valoctocogene roxaparvovec.12
After decades of research, gene therapy is on the cusp of widespread use once some of the common obstacles are addressed. Some of the most prominent problems associated with hemophilia A gene therapy are adverse reactions, cost, and the complexity of therapy.13
The most common adverse reaction associated with hemophilia adeno-associated virus-mediated gene therapy is the elevation of alanine aminotransferase levels, which is indicative of liver damage.13 This can be treated with a course of corticosteroids; however, the risk of this adverse reaction may result in a contraindication in patients with preexisting liver damage. Gene therapy is a radical innovation, and the initial costs for these medications will reflect that. In a 2018 survey of 256 hematologists/oncologists treating at least one patient with hemophilia, 27% thought that cost would be the biggest barrier to implementing gene therapy in hemophilia patients.14 When asked about their understanding of the mechanisms of action, technology, and latest data on gene therapy, 54% of the respondents had minimal to no confidence in their understanding of gene therapy and demonstrated knowledge deficits.14 As research on adeno-associated virus-mediated gene therapy continues, emphasis should be placed on continual medical education so that physicians understand and can properly treat patients with hemophilia.
The Pharmacist’s Role in Management
The role of the pharmacist in the management of hemophilia continues to expand as new products continue to be developed. Pharmacists play an important role in drug selection, disease management, and education. Education will be necessary not only for patients but also for the physicians who treat these patients. The pharmacist’s role is to be the drug expert on all disease states; therefore, as hemophilia medications become increasingly more targeted, pharmacists will need to stay up-to-date so that they can make optimal recommendations to healthcare providers. Physicians have expressed that they need a better understanding of new hemophilia treatments; therefore, pharmacists must be prepared to keep abreast of current research and contextualize the necessary information.
REFERENCES
1. CDC. What is hemophilia? www.cdc.gov/ncbddd/hemophilia/facts.html. Accessed July 7, 2020.
2. Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1-e47.
3. Moorehead PC, Chan AK, Lemyre B, et al. A practical guide to the management of the fetus and newborn with hemophilia. Clin Appl Thromb Hemost. 2018;24(suppl 9):29S-41S.
4. Stephensen D, Tait RC, Brodie N, et al. Changing patterns of bleeding in patients with severe haemophilia A. Haemophilia. 2009;15(6):1210-1214.
5. Kizilocak H, Young G. Diagnosis and treatment of hemophilia. Clin Adv Hematol Oncol. 2019;17(6):344-351.
6. Collins PW. Personalized prophylaxis. Haemophilia. 2012;18(suppl 4):131-135.
7. Jackson S, Santoro C, Gue D, et al. Clinical practice of personalized prophylaxis in hemophilia: illustrations of experiences and benefits from two continents. Clin Case Rep. 2019;7(4):689-694.
8. Ar MC, Balkan C, Kavakli K. Extended half-life coagulation factors: a new era in the management of hemophilia patients. Turk J Haematol. 2019;36(3):141-154.
9. FDA. Vaccines, blood & biologics. www.fda.gov/vaccines-blood-biologics. Accessed June 15, 2020.
10. Hemlibra (emicizumab-kxwh) package insert. South San Francisco, CA: Genentech, Inc; November 2017.
11. Castellone DD. Past, present and future options in the treatment of hemophilia A. Am Soc Clin Lab Sci. 2019;118.
12. BioMarin. BioMarin provides highlights of 4 years of clinical data from ongoing phase 1/2 study of valoctocogene roxaparvovec gene therapy for severe hemophilia A. https://investors.biomarin.com/2020-05-31-BioMarin-Provides-Highlights-of-4-Years-of-Clinical-Data-from-Ongoing-Phase-1-2-Study-of-Valoctocogene-Roxaparvovec-Gene-Therapy-for-Severe-Hemophilia-A. Accessed June 15, 2020.
13. Batty P, Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum Mol Genet. 2019;28(R1):R95-R101.
14. Hurst ST, Kadkhoda H, Warren C, Pasi KJ. Gene therapy in hemophilia: an assessment of hematologists’ knowledge gaps and attitudes. https://img.medscapestatic.com/pi/edu/qrcode/posters/gene-therapy-in-hemophilia-hem-knowledge-gaps-and-attitudes.pdf. Accessed June 15, 2020.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



