Health care providers often encounter patients who regard generic medications as being less safe and effective and of poorer quality than branded equivalents. Lower pricing, however, does not indicate an inferior product. Pharmacists must be fully conversant with the approval process for generic drugs so that they understand the similarities and differences between generic and branded products and can address patients’ concerns. This review examines the history of generic-drug legislation, outlines current testing methods required to determine equivalency of generic and brand-name products, discusses controversies surrounding generic-drug approval, and provides an overview of generic-drug references.
The Hatch-Waxman Act and the ANDA
In the past, the FDA required generic- drug manufacturers to perform the same costly and time-consuming preclinical and clinical trials (i.e., phase I, II, and III studies) as were conducted for the original brand-name drug in order to prove the generic product’s clinical efficacy and safety. The Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the Hatch-Waxman Act, streamlined criteria for FDA approval, allowing a manufacturer to demonstrate only that its product does not differ significantly from the reference product (FIGURE 1).1,2 The elimination of preclinical and clinical studies from the approval process for generic drugs relies on the fundamental assumption that if a new drug product can achieve the same rate and extent of absorption in the bloodstream as a drug with documented safety and efficacy, then the new product will produce an equal clinical effect.3
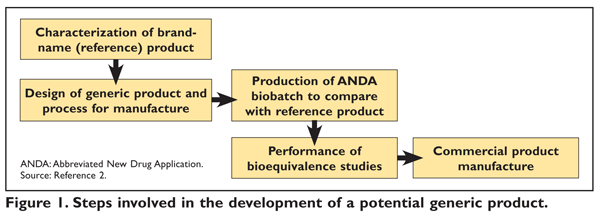
Manufacturers had little financial incentive to develop generic drugs before 1984, and generics accounted for less than 19% of the prescription-drug market.4 Because the Hatch-Waxman Act greatly reduced the time and expense required to develop and market generic drugs, generics now account for approximately 78% of drugs dispensed by retail pharmacies and long-term care facilities.4 The introduction of greater competition into the prescription-drug market ultimately translated into cost savings. The sale of generic drugs saved consumers and the U.S. health care system $158 billion in 2010 alone, and $931 billion over the past 10 years.5
The Hatch-Waxman Act established the Abbreviated New Drug Application (ANDA) for generic medications (TABLE 1).1,6 Generic-drug manufacturers file ANDAs through the FDA’s Center of Drug Evaluation and Research.

The Hatch-Waxman Act permits generic-drug manufacturers to conduct studies before the expiry of the innovator’s patent or market exclusivity.1 The generic manufacturer must certify its intentions with respect to each patent held by the innovator product (i.e., active ingredient, indication, formulation, and composition).1 Four certification options for submitting an ANDA exist under the Hatch-Waxman Act:
Paragraph I: The innovator product’s manufacturer has not filed patent information.
Paragraph II: The innovator product’s patent has already expired.
Paragraph III: The innovator product’s patent will expire on a future date.
Paragraph IV: The patent is
invalid or will not be infringed upon by the manufacture, use, or sale
of the generic product for which the ANDA has been submitted.1
The FDA may immediately approve ANDA requests certified under paragraph I or paragraph II. The generic manufacturer files for paragraph III certification if it has no intention of marketing its product until patent expiration, and ANDA approval is effective on the expiration date. The generic manufacturer must notify the patent holder if an ANDA is submitted under paragraph IV certification. The innovator company has 45 days to bring patent-infringement litigation against the generic manufacturer.1 A legal motion by the innovator company prohibits ANDA approval until either 30 months have elapsed or the court rules that the patent is invalid or would not be infringed upon by approval of the generic.1 The FDA grants 180 days of market exclusivity to the first company to successfully apply to market a generic version of the innovator drug under paragraph IV.1 Although federal law dictates that the FDA approve, tentatively approve, or disapprove an ANDA within 180 days of receipt, the FDA typically spends a median of 17 months reviewing an ANDA before making a decision.7
In exchange for the abbreviated generic-drug approval provisions, the Hatch-Waxman Act compensates brand-name manufacturers by offering up to 5 additional years of patent protection to account for time lost to clinical testing and regulatory review.1 A patent normally has a term of 20 years from the application date, but the effective patent term is usually far less than this because patents are often granted before the drug is marketed. The total patent life with the Hatch-Waxman patent extension cannot exceed 14 years from the product-approval date.1
Bioequivalence Studies
In the ANDA, the generic manufacturer must establish that its product is pharmaceutically equivalent to the branded product—i.e., the active ingredient(s), strength, dosage form, route of administration, and intended use are identical.1,8 Although excipients (e.g., binding agents, preservatives, flavorants, dyes) may differ, they must exist in similar proportion to the active ingredient(s) in the reference product.1,8 U.S. trademark law prohibits the generic product from physically resembling the brand-name product.9 Generic products must meet batch requirements for identity, strength, purity, and quality and must be manufactured according to the FDA’s Current Good Manufacturing Practice regulations.1,8 Although there is currently a disparity in inspection rates for foreign and domestic generic-drug facilities (which has created an incentive for companies to manufacture generic drugs abroad), the FDA aims to achieve uniform inspection coverage by 2017.9 The enactment of the Generic Drug User Fee Amendments in 2012 has authorized the FDA to collect up to $229 million annually in user fees from the generic-drug industry.9 Higher fees will be charged for foreign facilities.9 The FDA will allocate a portion of these funds to the inspection of facilities once every 2 years, according to a risk-based approach.9
A generic-drug manufacturer must also prove bioequivalence of the generic to the innovator drug.8,10 Bioequivalent products do not differ significantly in the rate or extent of absorption at the site of action.11 Most manufacturers compare the pharmacokinetic parameters of the generic drug and the brand-name drug to support a claim of bioequivalence, a method favored by the FDA.11 Alternative approaches, in order of FDA preference, include pharmacodynamic, comparative clinical, and in vitro studies.11 Drugs that are both pharmaceutically equivalent and bioequivalent meet the criteria for therapeutic equivalence and are interchangeable.11
A generic ANDA applicant typically conducts a two-way crossover study in a small group of healthy volunteers under standardized conditions.11,12 The number of subjects should be large enough to detect a 20% difference in the parameters with 80% certainty.3 The FDA recommends a minimum of 12 subjects, but most studies include 24 to 36.3 The FDA’s preference is that each drug be administered in single doses, since this method has been shown to be more sensitive than multiple-dose administration in assessing the release of active ingredient into the bloodstream.11 Bioequivalence studies routinely involve the highest strength of a drug product; studies are waived for lower-strength products as long as the active ingredient(s) and excipients are proportionally similar to those of the highest-strength product.3,11
In pharmacokinetic models, the peak drug concentration (Cmax) and the AUC characterize absorption rate and absorption extent, respectively (FIGURE 2).3,11,13 The FDA permits up to 20% variation between brand-name and generic drugs, noting that a ±20% difference in blood concentration for most drugs would not be clinically significant.3,11 Generic manufacturers perform two one-sided statistical tests on pharmacokinetic data.12 The statistical analyses determine whether the mean values for pharmacokinetic parameters of the test (generic) and reference products are comparable. The first statistical test verifies that the mean response of the generic drug is not more than 20% less than that of the innovator product, numerically expressed as a limit of 80% on the test mean/reference mean. The second test verifies that the average response of the innovator product is not more than 20% less than that of the generic drug, numerically expressed as a limit of 80% on the test mean/reference mean. Since all data are expressed in ratios of the test mean to the reference mean, the limit of the second statistical test becomes 125% (the reciprocal of 80%).3 Statisticians calculate a 90% confidence interval (CI) for the ratio of the test and reference products. The 90% CI must fall entirely within the 80% to 125% boundary.3,12
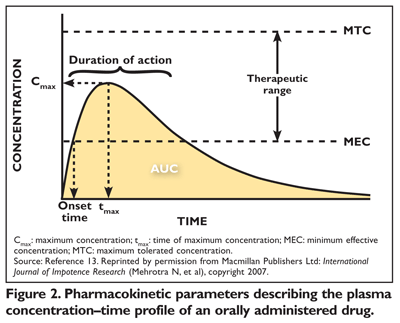
Critics often falsely interpret the width of the 80% to 125% limits to mean that the FDA permits a difference in Cmax and AUC between the generic and brand-name products by as much as 45%. In fact, the two drugs would almost certainly fail bioequivalence studies if such a large absolute difference between test and reference pharmacokinetic parameters existed. When the means of the Cmax and/or AUC of the two products differ by more than 12% to 13%, the generic product will likely fail to meet the 80% to 125% bioequivalence requirement.3
The FDA’s Office of Generic Drugs has conducted three surveys to quantify pharmacokinetic differences between generic and brand-name products.14-16 The largest survey evaluated 2,070 bioequivalence studies of generic drugs approved by the FDA from 1996 to 2007.16 The average difference between generic and innovator products was 4.35% and 3.56% for Cmax and AUC, respectively.16 Additionally, for nearly 98% of the bioequivalence studies reviewed, the AUC of the generic differed from that of the innovator product by less than 10%.16
The FDA is primarily concerned with protecting patients from approved drug products that are not bioequivalent. With the two one-sided tests method performed at 0.05 significance, there is no more than a 5% chance that a generic that is not truly equivalent to the innovator product will be approved.12
Controversies in FDA Bioequivalence Testing
Unresolved concerns surrounding bioequivalence undermine patients’ and health care providers’ confidence in making generic substitutions. Bioequivalence studies do not assess clinical or surrogate markers that directly correlate with efficacy and/or toxicity (e.g., thyroid-stimulating hormone [TSH], seizures, transplant rejection, international normalized ratio).17 In addition, manufacturers do not undertake comparative studies against other generic products with the same active ingredient, yet the FDA maintains that all approved generic medications are bioequivalent.17 Brand-name formulations must demonstrate pharmacokinetics, efficacy, and tolerability in both healthy patients and the drug’s target population, yet generic drugs demonstrate their bioequivalence to the brand-name products in homogeneous groups of normal and healthy subjects only.17
Several medical associations have released position statements opposing automatic generic substitution for brand-name narrow therapeutic index (NTI) drugs, since minor changes in blood concentrations can result in profound changes in efficacy or toxicity.18-21 For products with an NTI, European standards have restricted the acceptance interval for the Cmax and AUC to 90% to 111.11%, but they do not define criteria for classifying NTI drugs.22 The FDA has generally rejected the drug-by-drug approach in favor of a single set of bioequivalence standards. However, in 2010, the FDA began developing individualized study recommendations for certain products; most of these recommendations are currently in draft status and available for public comment.23
Levothyroxine, a synthetic form of the thyroid hormone thyroxine, is a classic example of why the FDA’s “one-size-fits-all” method of determining bioequivalence gives rise to concern. Even a minimal change in levothyroxine concentration may generate highly significant clinical results, especially in the elderly, pregnant patients, and those with cardiovascular disease.18 For this reason, multiple dosages of levothyroxine exist (e.g., there is only a 9% difference between 137 mcg and 150 mcg). In addition, measurements of levothyroxine levels do not account for patients’ endogenous baseline levels.18 An American Association of Clinical Endocrinologists statement recommends checking TSH levels 6 weeks after any change in medication (e.g., from brand to generic, from one generic to another generic).18
Despite the concerns over lack of true interchangeability, evidence of differences in clinical effectiveness between brand and generic products is unreliable. The strongest evidence for antiepileptic drugs (i.e., small prospective studies) suggests that brand-to-generic substitution likely is not problematic, although lesser-quality studies indicate that switching may result in increased utilization of health care resources.24 A systematic review and meta-analysis of 38 randomized, controlled trials compared generic cardiovascular drugs with their brand-name counterparts.24 Even among study drugs with an NTI (i.e., antiarrhythmic agents and warfarin), there was no evidence of clinical superiority for brand-name products.15
Orange Book and Drugs@FDA
The FDA publication Approved Drug Products With Therapeutic Equivalence Evaluations, also known as the Orange Book, lists all approved drug products.7 The FDA updates its online version of the Orange Book (www.accessdata.fda.gov/scripts/cder/ob/default.cfm) every few weeks.8 The Orange Book database may be searched by active ingredient or proprietary (brand) name.8 Listings include dosage form, route of administration, available strengths, drug manufacturer, and approval date.15 The Orange Book provides a therapeutic equivalence rating for all generic drugs and for the reference listed drug (RLD), which is the product against which an ANDA applicant tests the generic drug in order to gain FDA approval. Products with no known or suspected bioequivalence problems are designated AA (conventional dosage form), AN (aerosolized product), AO (injectable oil solution), AP (injectable aqueous solution), or AT (topical product).15 Products with known bioequivalence problems that have been resolved by adequate in vivo and/or in vitro evidence supporting bioequivalence are designated AB.8 A number designation follows the two-digit code when more than one RLD exists with the same active ingredient (e.g., AB1 and AB2).8 An AB1-rated RLD and an AB1-rated generic product—but not an AB2-rated generic product—are therapeutically equivalent and suitable for product substitution.
The FDA also maintains Drugs@FDA (www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm), a catalogue of FDA-approved products that is searchable by active ingredient, brand name, or application number. Links to label information, approval history, and regulatory reviews are provided.
Conclusion
The generic-drug market has expanded remarkably since the passage of the Hatch-Waxman Act in 1984, and the number of generics continues to grow as blockbuster drugs come off patent through 2015. Substituting a generic for a branded medication does not mean a sacrifice in quality, since the FDA holds generic manufacturers to the same high standards as brand-name manufacturers. Generic drugs must be proven nearly equal to innovator products in terms of rate and extent of absorption, which the FDA uses as surrogates for clinical efficacy and safety. It is important for pharmacists to understand the generic-drug approval process so that patients’ questions can be answered and the most cost-effective product can be dispensed.
REFERENCES
1. Drug Price Competition and Patent Term Restoration Act of 1984. Public Law 98-417, 98 Stat. 1585-1605, September 1984.
2. Lionberger RA. FDA critical path initiatives: opportunities for generic drug development. AAPS J. 2008;10:103-109.
3. Malinowski HJ, Johnson SB. Bioavailability and bioequivalency testing. In: Troy DB, ed. Remington: The Science and Practice of Pharmacy. 21st ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1037-1046.
4. Savings From Generic Drug Use. GAO-12-371R. Washington, DC: Government Accountability Office; 2012:2.
5. Savings: An Economic Analysis of Generic Drug Usage in the U.S. Washington, DC: Generic Pharmaceutical Association; 2011:1.
6. Sherwood T. Generic drugs: overview
of ANDA review process. CDER Forum for International Drug Regulatory
Authorities. www.fda.gov/downloads/Drugs/NewsEvents/UCM167310.pdf.
Accessed March 30, 2013.
7. The Food and Drug Administration’s Generic Drug Review Process. Washington, DC: Office of Inspector General; 2008.
8. U.S. Department of Health and Human
Services. Approved products with therapeutic equivalence evaluations.
33rd ed.
www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm071436.pdf.
Accessed March 24, 2013.
9. FDA. Fact sheet: new user fees for
generic drugs will enhance Americans’ access to less expensive drugs
and generate major cost savings.
www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FDASIA/ucm310992.htm.
Accessed April 21, 2013.
10. FDA. Facts about generic drugs.
www.fda.gov/downloads/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/UnderstandingGenericDrugs/UCM219406.pdf.
Accessed March 24, 2013.
11. Center for Drug Evaluation and Research. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations. Rockville, MD: FDA; 2003.
12. Center for Drug Evaluation and Research. Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. Rockville, MD: FDA; 2001.
13. Mehrotra N, Gupta M, Kovar A,
Meibohm B. The role of pharmacokinetics and pharmacodynamics in
phosphodiesterase-5 inhibitor therapy. Int J Impot Res. 2007;19:253-264.
14. Nightingale SL, Morrison JC. Generic drugs and the prescribing physician. JAMA. 1987;258:1200-1204.
15. Kesselheim AS, Misono AS, Lee JL, et
al. Clinical equivalence of generic and brand-name drugs used in
cardiovascular disease: a systematic review and meta-analysis. JAMA. 2008;300:2514-2526.
16. Davit BM, Nwakama PE, Buehler GJ, et
al. Comparing generic and innovator drugs: a review of 12 years of
bioequivalence data from the United States Food and Drug
Administration. Ann Pharmacother. 2009;43:1583-1597.
17. Meredith P. Bioequivalence and other unresolved issues in generic drug substitution. Clin Ther. 2003;25:2875-2890.
18. American Association of Clinical
Endocrinologists. AACE, ATS, and ATA joint position statement on the
use and interchangeability of thyroxine products.
www.aace.com/files/position-statements/aace-tes-ata-thyroxineproducts.pdf.
Accessed April 29, 2013.
19. Epilepsy Foundation. Position on
switching of antiepileptic drugs.
www.epilepsyfoundation.org/getinvolved/advocacy/positionstatements/genedrev.cfm.
Accessed March 25, 2013.
20. American Epilepsy Society. The
substitution of different formulations of antiepileptic drugs for the
treatment of epilepsy.
www.aesnet.org/go/press-room/consensus-statements/drug-substitution.
Accessed March 25, 2013.
21. Alloway RR, Isaacs R, Lake K, et al.
Report of the American Society of Transplantation conference on
immunosuppressive drugs and the use of generic immunosuppressants. Am J Transplant. 2003;3:1211-1215.
22. Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. London, England: European Medicines Agency; 2010:16.
23. Center for Drug Evaluation and Research. Guidance for Industry: Bioequivalence Recommendations for Specific Products. Silver Spring, MD: FDA; 2010.
24. Yamada M, Welty TE. Generic
substitution of antiepileptic drugs: a systematic review of prospective
and retrospective studies. Ann Pharmacother. 2011;45:1406-1415.
To comment on this article, contact rdavidson@uspharmacist.com.



