In the elderly, an initial vague complaint of weakness may be attributed to age-related sarcopenia or periarticular muscle weakness associated with osteoarthritis, common conditions in this population. However, it is important for pharmacists to be aware of, and to question patients to elicit, the nuances associated with conditions seen in the elderly, so as not to overlook an opportunity to refer a person for further evaluation that can positively impact outcome.
Myasthenia gravis (MG), while rare, is a neuromuscular disease that is classified as chronic and autoimmune in nature and is characterized by varying degrees of skeletal muscle weakness. And while MG may affect any voluntary muscle, those that control eye and eyelid movement, facial expression, and swallowing are most frequently affected. The onset of the disorder may be sudden, and symptoms often are not immediately recognized as MG.1
The Latin and Greek origin of the term myasthenia gravis literally means “grave muscle weakness”; however, recognition, diagnosis, and current therapies have enabled most individuals with MG to achieve a normal life expectancy.1 MG occurs in both genders and in all ethnic groups.1 The estimated prevalence of MG is approximately 20 cases per 100,000 population, with the disease affecting twice as many women as men.2 The overall prevalence is approximately 150-200 per million.3 However, in older age groups—late-onset MG, with onset after 50—men are affected more often and the disease is often misdiagnosed.4-6 As a result, there is a bimodal distribution, with a female predominance in the second to third decades of life, and male predominance in the sixth to eighth decades.3,4,7 While MG is not inherited, occasionally the disease may occur in more than one member of the same family.
Fatigability: Neuromuscular Junction
MG produces autoantibody- and cell-mediated destruction of acetylcholine receptors (AChRs) at the neuromuscular junction of the skeletal muscles.8-11 Symptoms arise once the number of AChRs is reduced to approximately 30% of normal.9 This decrease in number of AChRs results in a characteristic episodic pattern of progressively reduced skeletal muscle strength and fatigability with repeated use, and recovery of muscle strength after a period of rest.8,9
The most commonly and most severely affected muscles are the ocular and bulbar muscles; most patients also develop some degree of fluctuating generalized weakness.7 Smooth and cardiac muscle are usually not affected by MG since their cholinergic receptors have a different antigenicity compared with skeletal muscle.9
Clinical Manifestations and Complications
Symptoms of MG are outlined in TABLE 1. The usual initial complaint is a specific muscle weakness as opposed to a generalized weakness.9 Extraocular muscle weakness or ptosis is present initially in half of all patients and occurs during the course of illness in 90% of individuals.9 MG remains exclusively ocular in only 16% of patients.9 Ocular complaints are more common during the first year and are the presenting symptom in half of all cases; often within 1 year, patients have generalized symptoms such as weakness or fatigue and one third of patients develop respiratory weakness, requiring mechanical ventilation.2,12 If generalized myasthenia is going to develop after ocular symptoms, it usually does so within the first 3 years.8 Some patients present with bulbar symptoms (e.g., altered voice, nasal regurgitation, choking, dysphagia). Of note, hand grip may alternate between weak and normal, neck muscles may become weak, and proximal limb weakness is common.8 Sensation and deep tendon reflexes are normal. Manifestations fluctuate in intensity over minutes, hours, and days.8

Myasthenic crisis, a severe generalized quadriparesis or life-threatening respiratory muscle weakness, occurs in about 15% to 20% of patients at least once in their life, often secondary to a supervening infection that reactivates the immune system.8 Once respiratory insufficiency manifests, respiratory failure may occur rapidly; patients in myasthenic crisis should undergo evaluation for an infectious trigger.8
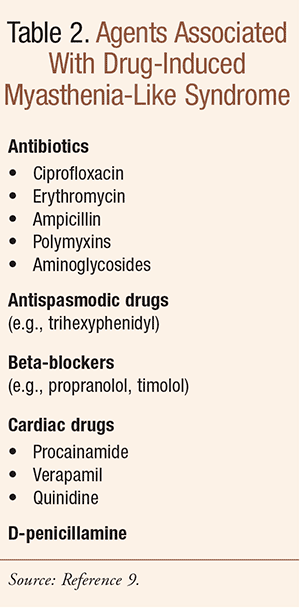
A variety of factors can trigger or worsen exacerbations in the elderly, including: 1) bright sunlight; 2) surgery; 3) immunization; 4) emotional stress; 5) intercurrent illness (e.g., viral infection); and 6) medications (e.g., aminoglycosides, ciprofloxacin, chloroquine, procaine, lithium, phenytoin, beta-blockers, procainamide, statins).9
Diagnosis and Differential Diagnoses
Diagnosis is by measurement of serum AChR antibody levels, electromyography, and in some cases IV edrophonium challenge, which briefly decreases the weakness; bedside testing is discussed in Reference 8. MG can mimic other diagnoses in elderly persons and vice versa. Differential diagnoses include myocardial infarction, pulmonary embolism, multiple sclerosis, amyotrophic lateral sclerosis, basilar artery thrombosis, brainstem gliomas, chronic myelogenous leukemia, and polymyositis, among others. Additionally, other problems should be considered, such as depression, and as mentioned above, a drug-induced myasthenia-like syndrome (TABLE 2), among others.9
Some conditions commonly accompany MG; therefore, after the diagnosis is confirmed, it is recommended that testing be done for thymic hyperplasia, thymomas, hyperthyroidism, and autoimmune disorders.8
Treatment
Patients with MG require close follow-up care in cooperation with the primary-care physician.9 While clinical features and management of MG in older adults are similar to that of younger patients, an increased risk of adverse effects of medications and comorbidities in the elderly requires even more careful monitoring.13,14 For most patients, the use of anticholinesterase drugs relieves symptoms; immunomodulating treatment slows disease progression and helps to relieve symptoms; TABLE 3 outlines pharmacologic therapy for MG.

Anticholinesterase drugs (e.g., pyridostigmine, neostigmine), which increase acetylcholine at the neuromuscular junction, form the basis of symptomatic treatment; however, they do not alter the underlying disease process and rarely relieve all symptoms.8 The disease may become refractory to these agents. Most patients eventually require treatment with corticosteroids or other immunosuppressants.15 Cholinergic crisis—muscular weakness secondary to an anticholinesterase drug dose that is too high—when mild, may be difficult to differentiate from worsening MG.8 Severe cases of cholinergic crisis, however, are unlike MG in that they produce increased lacrimation and salivation, tachycardia, and diarrhea.8 It is recommended that if patients deteriorate suddenly after responding well to treatment, respiratory support should be provided and anticholinesterase drugs stopped for several days.8 Anticholinesterase drugs can cause abdominal cramps and diarrhea, which are usually treated with oral atropine 0.4 to 0.6 mg (given with pyridostigmine or neostigmine) or propantheline 15 mg tid to qid.8 These interventions carry a significant anticholinergic burden in the elderly, including the increased risk for delirium in older adults.16 While bronchodilators may be helpful in treating bronchospasm associated with a cholinergic crisis, patients with respiratory failure require intubation and mechanical ventilation.8,9
Most patients eventually require treatment with corticosteroids or other immunosuppressants (TABLE 3). Prednisone is the most commonly used drug after pyridostigmine; since corticosteroid-associated osteoporosis is more likely in the elderly, it should be anticipated and prevented with bisphosphonates, calcium, and vitamin D.17
Azathioprine may be used as monotherapy and may be considered a corticosteroid-sparing agent, while cyclosporine is used mainly in severe MG that is not responding to prednisone and azathioprine.17 Adverse effects of these agents must be considered in assessing the overall clinical picture.9,18 Other treatments that may be beneficial—including drugs and thymectomy, which may be indicated—are also listed in TABLE 3.
Prognosis
Due to changes in treatment over the years, prognosis and mortality have also changed; mortality in the last 4 decades has seen a dramatic decrease from 75% to 4.5%.3 With treatment, muscle weakness can significantly improve in most patients, enabling them to lead normal or nearly normal lives.1 Additionally, some cases of MG may go into remission, and muscle weakness may disappear completely, enabling the discontinuation of medications. The goal of thymectomy is stable, long-lasting complete remission, which may occur in about 50% of patients who undergo this surgical procedure.1 In some cases, the weakness of MG may be severe enough to cause respiratory failure, requiring immediate emergency medical care.1
Conclusion
MG may be more common in the elderly. Symptoms worsen with muscle activity and lessen with rest; therefore, MG should be considered in patients with ptosis, diplopia, and muscle weakness after use of the affected muscle. While clinical features and management of MG in older adults are similar to those of younger patients, an increased risk of adverse effects of medications and comorbidities in the elderly requires even more careful monitoring.
REFERENCES
1. National Institutes of Health. National Institute of Neurological Disorders and Stroke. Myasthenia gravis fact sheet. September 2010. NIH Publication No. 10-768. www.ninds.nih.gov/disorders/myasthenia_gravis/detail_myasthenia_gravis.htm#3153_4. Accessed December 27, 2016.
2. Goldenberg WD. Emergent management of myasthenia gravis. Updated December 8, 2016. http://emedicine.medscape.com/article/793136-overview. Accessed December
27, 2016.
3. Li Y, Arora Y, Levin K. Myasthenia gravis: newer therapies offer sustained improvement. Cleve Clin J Med. 2013;80(11):711-721.
4. Spillane J, Higham E, Kullmann DM. Myasthenia gravis. BMJ. 2012. 345:e8497.
5. Mantegazza R, Baggi F, Antozzi C, et al. Myasthenia gravis: epidemiological data and prognostic factors. Ann NY Acad Sci. 2003;998:413-423.
6. Aarli JA. Late-onset myasthenia gravis: a changing scene. Arch Neurol. 1999;56:25-27.
7. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37(2):141-149.
8. Rubin, M. Myasthenia gravis. Merck Manual.com. Last full review/revision September 2016. www.merckmanuals.com/professional/neurologic-disorders/peripheral-nervous-system-and-motor-unit-disorders/myasthenia-gravis. Accessed November 14, 2016.
9. Shah AK. Myasthenia gravis. Medscape.com. Updated March 23, 2016. http://emedicine.medscape.com/article/1171206 medication#showall. Accessed November 21, 2016.
10. Vincent A, Newsom-Davis J. Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psych. 1985;48:1246-1252.
11. Nicolle MW. Myasthenia gravis. Neurologist. 2002;8:2-21.
12. Gwathmey K, Balogun RA, Burns T. Neurologic indications for therapeutic plasma exchange: an update. J Clin Apher. 2011;26(5):261-268.
13. Aragones JM, Bolibar I, Bonfill X. Myasthenia gravis: a higher than expected incidence in the elderly. Neurology. 2003;60:1024-1026.
14. Vincent A. Clover L, Buckley C. Evidence of underdiagnosis of myasthenia gravis in older people. J Neurol Neurosurg Psychiatry. 2003;74(8):1105-1108.
15. Bhanushali MJ, Wuu J, Benatar M. Treatment of ocular symptoms in myasthenia gravis. Neurology. 2008;71:1335-1341.
16. Reuben DB, Herr KA, Pacala JT, et al. Geriatrics at Your Fingertips: 2016. 18th ed. New York, NY: The American Geriatrics Society; 2016:67.
17. Doherty TJ, Nicolle MW. Neuromuscular disorders. In: Fillit HM, Rockwood K, Woodhouse K, eds. Brocklehurst’s Textbook of Geriatric Medicine and Gerontology. 7th ed. Philadelphia, PA: Saunders Elsevier; 2010:530-532.
18. Semla TP, Beizer JL, Higbee MD. Geriatric Dosage Handbook. 20th ed. Hudson, OH: Lexicomp; 2015:124-126.
19. Epocrates.com. Epocrates Plus Version 15.12.1. Updated December 16, 2016.
20. Tuteja S. Immunosuppressants. In: Whalen K, ed. Pharmacology. 6th ed. Philadelphia, PA: Wolters Kluwer; 2015:619-629.
To comment on this article, contact rdavidson@uspharmacist.com.


