US Pharm. 2016;41(12):16-19.
For nearly five decades, pharmacists have been dispensing generic drugs within the confines of existing laws. The recent emergence of biosimilars in the U.S. market begs for an understanding of generic equivalency regulations and how biosimilar medications may fit into existing laws or what changes may be necessary to allow pharmacists to dispense these new entities, with the goal of saving millions or even billions of dollars in the process.
Generic Drugs
Generics that are interchangeable with the brand-name or prescribed product must be therapeutically, biologically, and chemically equivalent. Drugs that are therapeutically the same or at least very close to the same as the brand-name drug but are not biologically and chemically equivalent cannot be interchanged generically. As an example, consider erythromycin base and erythromycin ethylsuccinate. These two agents may be therapeutically equivalent, but they are neither bioequivalent nor chemically identical and thus cannot be generically substituted for each other.1
Biosimilars are biological products that contain the same active molecule as the brand-name agent (reference product) but are not chemically or biologically identical and thus would not be interchangeable without special considerations.2,3 Brand-name drugs and their generic equivalents are considered small molecules, while biologics and biosimilars are generally large molecules.4
Biologics Price Competition and Innovation (BPCI) Act
The Affordable Care Act was signed into law on March 23, 2010.2,3 This complex law contains, among many other provisions, the BPCI Act. The goal of the BPCI Act is much like the Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act).5 Passage of the Hatch-Waxman Act created an abbreviated approval process for small-molecule products based predominantly on the ability of generic manufacturers to demonstrate bioequivalence with the reference product. The primary purpose of the Hatch-Waxman Act was to expedite the availability of generic drugs so as to increase price competition between branded reference products and less costly generics. This act has been widely successful at creating the intended competition, which has resulted in significant cost savings for small-molecule drugs.5 Likewise, the BPCI Act establishes an abbreviated pathway for the approval of biological products that are “highly similar” (i.e., biosimilar) to or interchangeable with the reference biological product.4
What Are Biosimilar Agents?
The U.S. government defines a biological product as “any virus, therapeutic serum, toxin, antitoxin, or analogous product applicable to the prevention, treatment, or cure of diseases or injuries of man.”6 Biosimilars are a type of biological product approved by the FDA because they are “highly similar” to an already FDA-approved biological product, known as the reference product, and have been shown to have no clinically meaningful differences from the reference product. Minor differences in clinically inactive components are allowed, but no clinically meaningful differences between the biosimilar and the reference product it was compared to in terms of safety, purity, and potency are permitted.2
What Are Interchangeable Products?
Interchangeable products are both biosimilar to an FDA-approved reference product and can be expected to produce the same clinical result in patients as the reference product. An interchangeable product may be substituted for the reference product without the intervention of the healthcare provider who prescribed the reference product. In addition, for a biological product that is administered more than once to an individual, the risk in terms of safety or efficacy of alternating or switching between the biological product and the reference product will not be greater than the risk of using the reference product without alternating or switching.2
The Purple Book
Much the way the Orange Book indicates the bioequivalence of generic drugs,1 the Purple Book lists biological products, including any biosimilar and interchangeable biological products, licensed by the FDA under the Public Health Service (PHS) Act.7 The Purple Book includes the date a biological product was licensed under §351(a) of the PHS Act and whether the FDA evaluated the biological product for reference product exclusivity under §351(k)(7) of the PHS Act. The Purple Book, in addition to the date licensed, also includes whether a biological product licensed under §351(k) of the PHS Act has been determined by the FDA to be biosimilar to or interchangeable with a reference biological product.7
State Laws
For decades, states have regulated the use of brand-name and generic prescription drugs through statutes and agency or board rules. These state actions include when and how generics may be substituted for brand-name prescription drugs by pharmacists or others. Regulating biological products raises new issues for both state and federal policymakers. Because of their complexity, biologics are much more difficult to replicate than the chemically produced generics for other drugs. As a result, truly identical generic versions of biologics are virtually impossible to produce. However, once patents expire for the existing brand-name biological products, biosimilar medications can be produced, which is an occurrence that raises regulatory issues in the states.8
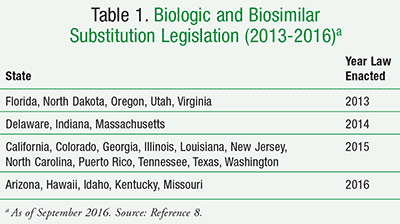
There is concern that traditional statutes regulating generic drugs may be misapplied to new products that are not identical. This has led to a recent move to amend older state laws to address the medical and chemical characteristics of these biologics, as well as any future generic-style “follow-on biologics” or biosimilars. In the past 4 years, a cumulative total of at least 36 states have considered legislation establishing state standards for substitution of a biosimilar prescription product to replace an original biological product, and 23 states (plus Puerto Rico) have passed some sort of legislation (TABLE 1).8
Naming of Biosimilar Agents
The current naming convention was adopted after release of the FDA draft guidance on biosimilar naming in August 2015.9 This draft guidance recommends that all previously approved, currently under review, and future biosimilars as well as current and existing reference biologics be named based on a nonproprietary name with an assigned hyphenated suffix. The FDA’s draft guidance reported the intent to designate each approved biologic with a nonproprietary name, including a randomly assigned 4-letter suffix that is devoid of meaning. The current draft guidance does not detail the naming requirements for any future biologics that are approved as an interchangeable biologic from the FDA.9
The FDA’s draft guidance stresses the need for improved pharmacovigilance and to clearly differentiate all products that are not deemed interchangeable as the rationale for assigning unique suffixes to each unique biologic.9 In a study of pharmacists’ preferences on the naming of biosimilars, those supporting the use of a unique biologic qualifier or unique overall name for biosimilars cite the importance of using the unique product names to better track patient- and healthcare-provider–reported adverse events.5 Supporters state that using the same nonproprietary name for biosimilars could result in confusion in the reporting process and make pharmacovigilance studies more difficult and inaccurate.
Opponents cite the presence of unique identifiers, such as National Drug Code (NDC) and lot numbers, as reasons why assigning a suffix or other biologic qualifier or unique product name is not necessary. Opponents also cite the potential to create confusion among providers and patients. This study concluded that “data from the FDA Adverse Event Reporting System have suggested an adverse-reporting trend toward continued adverse reports being assigned to the brand-name reference product even after a shift in market to primarily using generic products.”5
The European Experience
Biosimilar agents have been on the market in European countries for over 10 years.6 The European Medicines Agency (EMA) oversees the safety, scientific evaluation, and marketing authorization of human and veterinary medications within the European Union (EU).4 A centralized process allows pharmaceutical manufacturers to submit a single application to the EMA, that if accepted, is valid for all EU members.4 The EMA developed its first guideline on “similar biological medicinal products [biosimilars]” in June 2004, which was published in October 2005; the first biosimilar was approved in April 2006.4,6 To date, there are approximately 20 biosimilars that have either received marketing approval or are in the approval pipeline in Europe.4
The EMA’s evaluation and approval process for biosimilars is comparable to what the FDA employs.4 The EMA requires that pharmaceutical manufacturers conduct well-performed studies with results that clearly demonstrate that the proposed biosimilar medication has no significant differences in quality, safety, or efficacy from the reference product.4.6
Biosimilars in the U.S.
In March 2013, the U.S. held its first formal meeting between the FDA and biosimilar/biological product manufacturers, but it was not until March 2015 that the first biosimilar agent received FDA approval.4 Zarxio (filgrastim-sndz) is a biosimilar to Neupogen (filgrastim); both are indicated for the treatment
of neutropenia in cancer patients undergoing chemotherapy.4,10 At present, there are only four FDA-approved biosimilars available (TABLE 2), although many others are in development.10 None of these agents is currently approved as an interchangeable product.

Most recently, Amgen’s Amjevita (adalimumab-atto), a biosimilar version of AbbVie’s anti-inflammatory drug Humira (adalimumab), was approved by the FDA to treat conditions including psoriatic arthritis, rheumatoid arthritis, psoriasis, and Crohn’s disease.11 The agent is the fourth biosimilar approved by the agency.11 Humira was the second-best-selling prescription drug in 2015 at $14 billion,12 while sofosbuvir, the active ingredient used for the treatment of hepatitis C, was number-one (through the two drugs Harvoni and Sovaldi, sofosbuvir brought in combined sales of almost $19 billion).13
Overall, biological products constitute a large portion of prescription-drug spending. It is hoped that the introduction of biosimilars will help reduce costs as generic drugs have. For example, the wholesale list price for Zarxio has been set at 15% below that of its reference product, Neupogen.10 It has been estimated by Express Scripts that biosimilars could save the U.S. healthcare system as much as $250 billion over the next 10 years.10 However, it is too soon to tell if the expected cost savings from biosimilars will be realized.12
“The biosimilar pathway is still a new frontier and one that we expect will enhance access to treatment for patients with serious medical conditions,” said Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research.11
The Pharmacist’s Role
As the relatively high-cost biologics and biosimilars start to move out from specialty practices into community pharmacies, pharmacists will play a vital role in helping to reduce healthcare costs. Pharmacists are well equipped to provide information to patients, consumers, and other healthcare providers while ensuring that the biosimilars that are dispensed are accurate as prescribed, safe and effective, and within the legal requirements of state and federal laws.
REFERENCES
1. FDA. Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations. www.accessdata.fda.gov/scripts/cder/ob. Accessed September 30, 2016.
2. FDA. Information for healthcare professionals (biosimilars). May 10, 2016. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241719.htm. Accessed September 24, 2016.
3. FDA. Information on biosimilars. May 10, 2016. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars. Accessed September 30, 2016.
4. Niemann C, Marshall JC, Van Hook L. Law: primer on biosimilar agents. Drug Topics. January 2016. http://drugtopics.modernmedicine.com/sites/default/files/images/DrugTopics/UCONN/2016-01-CE.pdf. Accessed September 29, 2016.
5. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926. www.jmcp.org/doi/full/10.18553/jmcp.2016. 22.8.919. Accessed September 29, 2016.
6. American Society of Health-System Pharmacists. A health-system pharmacist’s guide to biosimilars: regulatory, scientific, and practical considerations. Published 2013. www.ashpadvantagemedia.com/downloads/biosimcentral_guidelines.pdf. Accessed September 29, 2016.
7. FDA. Purple Book: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. Accessed September 30, 2016.
8. Cauchi R. State laws and legislation related to biologic medications and substitution of biosimilars. National Conference of State Legislators. September 16, 2016. www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed September 29, 2016.
9. FDA. Nonproprietary naming of biological products. Guidance for industry (draft guidance). August 2015. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM459987.pdf. Accessed November 17, 2016.
10. Panesar K. Biosimilars: current approvals and pipeline agents. US Pharm. 2016;41(10):26-29. www.uspharmacist.com/article/biosimilars-current-approvals-and-pipeline-agents. Accessed November 17, 2016.
11. FDA approves Amjevita, a biosimilar to Humira. FDA news release. September 23, 2016. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed November 17, 2016.
12. Minaya E. FDA approves Amgen’s biosimilar version of Humira. Wall Street Journal. September 23, 2016. www.wsj.com/articles/fda-approves-amgens-biosimilar-version-of-humira-1474669560. Accessed September 30, 2016.
13. Top drugs by sales revenue in 2015: who sold the biggest blockbuster drugs? PharmaCompass. March 10, 2016. www.pharmacompass.com/radio-compass-blog/top-drugs-by-sales-revenue-in-2015-who-sold-the-biggest-blockbuster-drugs. Accessed September 30, 2016.
To comment on this article, contact rdavidson@uspharmacist.com.



