US Pharm. 2021;46(11):HS1-HS6.
ABSTRACT: Euglycemic diabetic ketoacidosis (EDKA) is a rare, acute, life-threatening emergency that is characterized by euglycemia, metabolic acidosis, and ketoacidosis. Unlike DKA, the diagnosis of EDKA is often overlooked because of the absence of hyperglycemia. The mechanism behind EDKA involves a general state of starvation that results in ketosis while normoglycemia is maintained. EDKA may be associated with precipitating factors, including sodium-glucose cotransporter 2 inhibitors, starvation, pregnancy, alcohol use, surgery, and drug-induced intoxication. A stepwise approach is used in the management of EDKA. Pharmacists can assist the medical team in preventing EDKA and can play a role in the management of EDKA.
The management of type 1 and type 2 diabetes mellitus (T1DM, T2DM) has evolved with the availability of various antidiabetic agents.1 Biguanides—most notably metformin—have been used in conjunction with sulfonylureas, thiazolidinediones, glucagon-like peptide-1 receptor agonists, and dipeptidyl peptidase-4 inhibitors to treat T2DM.2 Sodium-glucose cotransporter 2 inhibitors (SGLT2i) are the newest class of oral diabetes medications.3 Besides maintaining glycemic control, SGLT2i also demonstrate cardiovascular and renal benefits. Therefore, SGLT2i have quickly become a mainstay of therapy in diabetes management.4 However, euglycemic diabetic ketoacidosis (EDKA) has emerged as a rare but serious adverse effect associated with SGLT2i. The risk of EDKA from SGLT2i is increased sevenfold in patients with T2DM, with an estimated overall incidence of approximately 0.1%.5,6 Pharmacists should be well versed in EDKA and its precipitating factors in order to educate patients and clinicians on the signs and management of EDKA in patients taking SGLT2i.
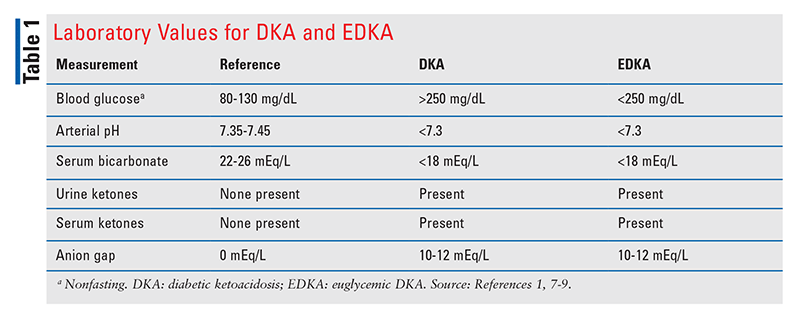
DKA, a life-threatening complication of diabetes, is characterized by the triad of hyperglycemia, metabolic acidosis, and ketoacidosis. Left untreated, DKA can lead to severe dehydration, cerebral edema, and coma. Hyperglycemia is a key criterion in the diagnosis of DKA; however, approximately 2.6% to 3.2% of DKA admissions are cases of EDKA, in which metabolic acidosis and ketoacidosis are accompanied by euglycemia. See TABLE 1.1,7-9
Pathophysiology
The mechanism behind EDKA involves a general state of starvation that results in ketosis while normoglycemia is maintained.1 Simply put, EDKA is DKA in which normal glucose concentrations are present. Diabetic patients, especially those on insulin, may not recognize symptoms as DKA because the serum glucose is not elevated.1 In the presence of a carbohydrate deficit due to precipitating factors, there is a reduction in serum insulin and an excess of glucagon, epinephrine, and cortisol.9 The increase in glucagon and decrease in insulin shift the metabolism toward lipolysis, an increase in free fatty acids (FFAs), and ketoacidosis.1 Ketone bodies—including acetoacetic acid, beta-hydroxybutyric acid, and acetone—are released, resulting in metabolic acidosis.1 Other factors that can contribute to EDKA are 1) the decrease in hepatic glucose production during a fasting state when glycogen stores are already depleted and 2) the increased urinary excretion of glucose.1
EDKA Precipitating Factors
SGLT2i: In the United States, SGLT2i (canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin) are currently approved to treat only T2DM, not T1DM. SGLT2i have demonstrated additional benefits for cardiovascular disease, chronic kidney disease, and heart failure. In fact, dapagliflozin is the only member of this class that has been approved for heart failure treatment regardless of a T2DM diagnosis.10 In Europe, dapagliflozin is approved for use in T1DM patients with a BMI of 27 kg/m2 or higher.10 The FDA has yet to approve SGLT2i as an adjunct to insulin in T1DM patients, owing to the increased risk of fatal DKA.11 Off-label use of SGLT2i has led to several documented cases of EDKA.10 SGLT2i prevent reabsorption of glucose by blocking sodium-dependent glucose transporter 2 in the proximal convoluted tubule. This mechanism enhances urinary excretion of glucose, resulting in lower plasma glucose concentrations.5 Low plasma glucose concentrations, in turn, create a carbohydrate deficit and volume depletion, stimulating glucagon secretion and suppressing insulin production. SGLT2i also directly stimulate the release of glucagon from the pancreas. The resulting lipolysis and ketogenesis are further worsened by the ability of SGLT2i to increase ketone reabsorption. The use of SGLT2i, combined with other precipitating factors, such as starvation (low-caloric diet), pregnancy, alcohol, acute infection, and procedures, can exacerbate EDKA. Reduced serum glucose, reduced insulin, increased glucagon release, and reduced clearance of ketones are contributors to EDKA in patients taking SGLT2i. Nondiabetic patients who are initiated on these medications may be at risk for developing EDKA. A study involving the administration of SGLT2i in healthy nondiabetic rats found that when they were exposed to volume-depleting stress, the rats developed EDKA.12
On May 15, 2015, the FDA issued a warning that SGLT2i may cause DKA in T1DM and T2DM patients.13 In the FDA Adverse Event Reporting System database, 73 cases of ketoacidosis in patients with T1DM or T2DM were reported from March 2013 to May 2015.5,13 All patients required hospitalization or treatment in an emergency department (ED).13 Treatment in these cases was delayed until the presentation of normal blood glucose (BG) concentrations. On March 19, 2020, the FDA issued an update regarding the risk of EDKA development after surgery in patients on SGLT2i.13 Canagliflozin, dapagliflozin, and empagliflozin should be stopped at least 3 days prior to a procedure, and ertugliflozin should be stopped at least 4 days prior.3,13
Low Caloric Intake or Starvation: A low-caloric diet or starvation in a patient with or without diabetes can trigger EDKA.14 A reduction in dietary uptake, especially carbohydrates, leads to the use of FFAs and ketones as the primary source of energy.8 Lipolysis and ketogenesis occur during prolonged fasting and lead to ketoacidosis. Glycogen stores are also entirely depleted, resulting in ketoacidosis with euglycemia. One case report described a 51-year-old female with no history of diabetes or excessive alcohol use who presented to the ED with altered general status, nausea, vomiting, deep asthenia, and articular pain after engaging in a 4-day restrictive diet followed by 48 hours of fasting.15 Her laboratory tests indicated a pH of 7.2, BG of 8 mmol/L (160 mg/dL), anion gap of 23 mmol/L, and positive serum lactate and ketone bodies. The patient recovered from ketoacidosis within 48 hours of glucose infusion without the need for insulin.15 In a similar case, a 24-year-old male without a history of diabetes or excessive alcohol use presented to the ED with shortness of breath, tachypnea, and sinus tachycardia after 3 days of fasting caused by nausea and vomiting.16 His laboratory tests indicated a pH of 7.09, BG of 6 mmol/L (106 mg/dL), high anion gap, normal lactate, and positive urine ketones. The patient recovered from ketoacidosis within 12 hours of being started on IV dextrose with no insulin.16
Pregnancy: Ketoacidosis in pregnancy is detrimental not only to the mother but also to the developing fetus, as it is linked to high fetal mortality rates.17 Pregnancy creates intrinsic physiological changes that predispose those with T1DM, T2DM, or gestational diabetes mellitus (GDM) to ketoacidosis.18 From 0.8% to 1.1% of DKA cases in pregnant women are EDKA.18 Pregnancy creates an insulin-deficient state because of placentally derived hormones, including glucagon, cortisol, and human placental lactogen, which are produced during periods of stress. Decreased insulin sensitivity is considered a physiological mechanism for delivering glucose to the developing fetus and placenta, which in turn reduces maternal BG concentrations.19 The insulin resistance increases with gestational age and may be exacerbated when coupled with prolonged fasting in the second and third trimesters. Maternal metabolism, as a result, is diverted to lipolysis for energy.19 One study discovered significant production of FFAs and beta-hydroxybutyrate after 12 hours of fasting in pregnant women in their third trimester, compared with nonpregnant women.20 Several cases of EDKA in pregnant women with T1DM, T2DM, or GDM have been reported in the third trimester (32-37 weeks’ gestation); patients had an average age of 30 years and BG of 110 mg/dL.17
Acute Onset of Infection: An acute onset of infection in diabetic and nondiabetic patients may also cause EDKA. One case report described a nondiabetic female with a known history of alcoholism who presented with metabolic acidosis and acute pancreatitis.21 The patient had not eaten for >1 week. The acidosis improved only after EDKA was suspected, insulin infusion dextrose was administered, and fluid/electrolyte abnormalities were addressed.21 Another case report involved a 76-year-old nondiabetic female with a history of low caloric intake and alcohol abuse who presented with septic shock due to acute obstructive cholangitis and EDKA.22 Sepsis can increase levels of counterregulatory hormones and insulin resistance, and these changes can induce ketogenesis in persons without diabetes. Septic shock can also induce renal dysfunction, leading to decreased excretion of ketones.
Drug-Induced Intoxication: Drug-induced intoxication, including that caused by cocaine, has been identified as a potential cause of EDKA. A case report described a 57-year-old female with T2DM who presented to the ED with altered mental status, nausea, vomiting, and abdominal pain.23 The patient was nonadherent to insulin monotherapy and reported poor oral intake. Laboratory results indicated urine positive for ketones and cocaine, pH of 7.02, and anion gap of 46.23 Cocaine has been reported to be a trigger for hyperglycemia because of its stimulatory effect on cortisol, epinephrine, and norepinephrine release from the adrenal gland.23,24 Conversely, cocaine can suppress the feeding centers in the central nervous system, resulting in a state of anorexia.24
Surgery: The American Association of Clinical Endocrinologists and the American College of Endocrinology recommend stopping SGLT2i at least 24 hours before an anticipated surgery, procedure, or stressful physical activity (e.g., marathon running).25 However, several case reports suggest that the effects of SGLT2i persist beyond five half-lives of elimination (2-3 days), with glucosuria and ketonemia present even 8 to 10 days after discontinuation.26 One case report described a 50-year-old female with T2DM who presented to the ED with constipation, fatigue, and reduced oral intake for 3 days prior to admission.26 The patient discontinued her regimen of metformin and dapagliflozin 2 days prior to admission. Laboratory results revealed acidosis with a pH of 7.34, elevated anion gap, beta-hydroxybutyrate, severe hypokalemia, hypophosphatemia, and acute kidney injury. Despite discontinuation of dapagliflozin 2 days prior to admission, glucosuria and ketoacidosis were seen for 8 days after the last use of dapagliflozin.26 In a review of nine cases of EDKA in both T1DM and T2DM patients, four patients in the T1DM subset had recurrent EDKA when restarted on SGLT2i and one patient presented with ketonuria 48 hours after discontinuation of canagliflozin.27 In a T2DM patient taking canagliflozin prior to a scheduled surgery, metabolic recovery did not occur until 6 days following the last dose of canagliflozin.27
Treatment
TABLE 2 outlines a stepwise approach to the management of EDKA.2,7,9,28-31

Step 1—Stop Inciting Agent, if Applicable: In the case of EDKA induced by SGLT2i or drug intoxication, the inciting agent(s) must be discontinued as soon as EDKA is diagnosed.1,7 An appropriate medication reconciliation is important to assist in establishing differential diagnoses including EDKA as well as helping determine optimal management.
Step 2—Start Fluid Replacement With Monitoring of Electrolytes and Ketones: Fluid resuscitation should be the focus of initial management in EDKA.28 Fluid loss due to EDKA can range from 6 L to 9 L, and rehydration is necessary for adequate tissue perfusion and resolution of metabolic abnormalities.28 The American Diabetes Association recommends 1 L/hour to 1.5 L/hour of normal saline or lactated Ringer’s solution during the first 1 to 2 hours of fluid resuscitation.28 Treatment with IV fluid supplementation should continue as appropriate based on patient factors until the anion gap closes and acidosis has resolved.28 Ketones and electrolytes should be monitored hourly and every 2 hours, respectively, until blood ketones are <0.6 mmol/L and electrolytes are stabilized.32
Step 3—Start Continuous Insulin Infusion: Despite the absence of hyperglycemia in EDKA, insulin plays an important role in the treatment and management of EDKA.7 Insulin suppresses the formation of ketones by promoting utilization of glucose via decreasing gluconeogenesis and glycogenolysis.28 Adequate fluid replacement should be followed by continuous insulin infusion, starting at a rate of 0.05 U/kg/hour to 0.1 U/kg/hour with serum potassium levels >3.3 mEq/L.1,2,33 Insulin causes intracellular movement of potassium into muscle cells. Therefore, if hypokalemia is present, insulin therapy should be delayed until potassium normalizes. Adequate monitoring of potassium levels should be conducted every 2 hours until electrolyte stabilization.32 Once EDKA has resolved, the patient may be started on SC long-acting insulin and premeal rapid-acting insulin to control BG. The insulin infusion should be continued for at least 1 hour after SC insulin is given.28
Step 4—Start Dextrose Administration: EDKA treatment requires that dextrose 5% (D5W) be added to fluids because of BG concentrations <250 mg/dL.1 Dextrose must be given to restore normal cellular utilization, resulting in enhanced clearance and reduced production of ketone bodies.29 The addition of D5W to fluids also prevents hypoglycemia by serving as an exogenous source of glucose in the setting of insulin utilization.1,29 If ketoacidosis persists despite administration of D5W, dextrose 10% may be used.9,30,33
The Pharmacist’s Role
Discussion With the Medical Team: EDKA can be a diagnostic challenge for clinicians owing to the absence of hyperglycemia from the otherwise typical presentation of DKA. Failure to recognize EDKA can result in delayed treatment, leading to serious complications such as cerebral edema and coma. The incidence of EDKA has grown with the introduction of SGLT2i, but this condition can also be seen in pregnancy, starvation, infection, and surgery. EDKA must be suspected in any patient presenting with metabolic acidosis and ketoacidosis, even in the presence of euglycemia. Pharmacists must educate the medical team regarding preventive measures, such as withholding SGLT2i treatment prior to a procedure as well as managing EDKA. Pharmacists can also play a key role in conducting comprehensive medication reconciliations and educating providers about the risk of EDKA in patients on SGLT2i therapy.
Discussion With the Patient: Patients may not recognize EDKA because of the normal BG concentration. Pharmacists must counsel patients on the presentation of EDKA, including nausea, vomiting, shortness of breath, lethargy, loss of appetite, fatigue, and abdominal pain.1 Patients with diabetes or with conditions predisposing them to EDKA should be educated about the signs and symptoms of ketoacidosis despite normal BG concentrations. To prevent EDKA, patients should be adherent to medications and consistently consume adequate meals throughout the day. Pharmacists must counsel patients about circumstances in which SGLT2i must be withheld, including prior to procedures, stressful physical activity (e.g., marathon running), acute illness, and low caloric intake.
Conclusion
EDKA is a high-risk yet obscure condition that can be challenging to diagnose and manage. As therapeutics experts, pharmacists are well suited to collaborate with other members of the healthcare team, as well as the patient, to identify risk factors for EDKA and to help appropriately manage this complex condition.
REFERENCES
1. Plewa MC, Bryant M, King-Thiele R. Euglycemic diabetic ketoacidosis. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; January 2021. www.ncbi.nlm.nih.gov/books/NBK554570/. Accessed July 22, 2021.
2. American Diabetes Association. Standards of medical care in diabetes—2021. https://care.diabetesjournals.org/content/diacare/suppl/2020/12/09/44.Supplement_1.DC1/DC_44_S1_final_copyright_stamped.pdf. Accessed August 16, 2021.
3. Invokana (canagliflozin) package insert. Titusville, NJ: Janssen Pharmaceuticals, Inc; March 2016.
4. Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013;1(2):140-151.
5. Blau JE, Tella SH, Taylor SI, Rother KI. Ketoacidosis associated with SGLT2 inhibitor treatment: analysis of FAERS data. Diabetes Metab Res Rev. 2017;33(8):e2924.
6. Erondu N, Desai M, Ways K, Meininger G. Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care. 2015;38(9):1680-1686.
7. Bonora BM, Avogaro A, Fadini GP. Euglycemic ketoacidosis. Curr Diab Rep. 2020;20(7):25.
8. Rawla P, Vellipuram AR, Bandaru SS, Pradeep Raj J. Euglycemic diabetic ketoacidosis: a diagnostic and therapeutic dilemma. Endocrinol Diabetes Metab Case Rep. 2017;2017:17-0081.
9. Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38(9):1638-1642.
10. Farxiga (dapagliflozin) package insert. Wilmington, DE: AstraZeneca Pharmaceuticals LP; May 2020.
11. Nathan DM. Adjunctive treatments for type 1 diabetes. N Engl J Med. 2017;377:2390-2391.
12. Perry RJ, Rabin-Court A, Song JD, et al. Dehydration and insulinopenia are necessary and sufficient for euglycemic ketoacidosis in SGLT2 inhibitor-treated rats. Nat Commun. 2019;10(1):548.
13. FDA. FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. www.fda.gov/files/drugs/published/Drug-Safety-Communication--FDA-warns-that-SGLT2-inhibitors-for-diabetes-may-result-in-a-serious-condition-of-too-much-acid-in-the-blood.pdf. Accessed August 16, 2021.
14. Burge MR, Garcia N, Qualls CR, Schade DS. Differential effects of fasting and dehydration in the pathogenesis of diabetic ketoacidosis. Metabolism. 2001;50(2):171-177.
15. Larroumet A, Camoin M, Foussard N, et al. Euglycemic ketoacidosis induced by therapeutic fasting in a non-diabetic patient. Nutrition. 2020;72:110668.
16. Owen D, Little S, Leach R, Wyncoll D. A patient with an unusual aetiology of a severe ketoacidosis. Intensive Care Med. 2008;34(5):971-972.
17. Jaber JF, Standley M, Reddy R. Euglycemic diabetic ketoacidosis in pregnancy: a case report and review of current literature. Case Rep Crit Care. 2019;2019:e8769714.
18. Chauhan SP, Perry KG, McLaughlin BN, et al. Diabetic ketoacidosis complicating pregnancy. J Perinatol. 1996;16(3 Pt 1):173-175.
19. Sinha N, Venkatram S, Diaz-Fuentes G. Starvation ketoacidosis: a cause of severe anion gap metabolic acidosis in pregnancy. Case Rep Crit Care. 2014;2014:906283.
20. Metzger B, Ravnikar V, Vileisis R, Freinkel N. “Accelerated starvation” and the skipped breakfast in late normal pregnancy. Lancet. 1982;1(8272):588-592.
21. Prater J, Chaiban JT. Euglycemic diabetic ketoacidosis with acute pancreatitis in a patient not known to have diabetes. AACE Clin Case Rep. 2015;1(2):e388-e391.
22. Nakamura K, Inokuchi R, Doi K, et al. Septic ketoacidosis. Intern Med. 2014;53(10):1071-1073.
23. Abu-Abed Abdin A, Hamza M, Khan MS, Ahmed A. Euglycemic diabetic ketoacidosis in a patient with cocaine intoxication. Case Rep Crit Care. 2016;2016:4275651.
24. Sarnyai Z, Dhabhar FS, McEwen BS, Kreek MJ. Neuroendocrine-related effects of long-term, ‘binge’ cocaine administration: diminished individual differences in stress-induced corticosterone response. Neuroendocrinology. 1998;68(5):334-344.
25. Handelsman Y, Henry RR, Bloomgarden ZT, et al. American Association of Clinical Endocrinologists and American College of Endocrinology position statement on the association of SGLT-2 inhibitors and diabetic ketoacidosis. Endocr Pract. 2016;22(6):753-762.
26. Pujara S, Ioachimescu A. Prolonged ketosis in a patient with euglycemic diabetic ketoacidosis secondary to dapagliflozin. J Investig Med High Impact Case Rep. 2017;5(2):2324709617710040.
27. Peters AL, Buschur EO, Buse JB, et al. Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care. 2015;38(9):1687-1693.
28. Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343.
29. Munro JF, Campbell IW, McCuish AC, Duncan LJ. Euglycaemic diabetic ketoacidosis. Br Med J. 1973;2(5866):578-580.
30. Diaz-Ramos A, Eilbert W, Marquez D. Euglycemic diabetic ketoacidosis associated with sodium-glucose cotransporter-2 inhibitor use: a case report and review of the literature. Int J Emerg Med. 2019;12(1):27.
31. Svart MV, Voss TS, Bayat M, et al. Rare presentations of ketoacidosis: diabetic ketoalkalosis and ketoacidosis secondary to fasting and muscular dystrophy. Clin Diabetes. 2015;33(1):37-39.
32. Muneer M, Akbar I. Acute metabolic emergencies in diabetes: DKA, HHS and EDKA. Adv Exp Med Biol. 2021;1307:85-114.
33. Nasa P, Chaudhary S, Shrivastava PK, Singh A. Euglycemic diabetic ketoacidosis: a missed diagnosis. World J Diabetes. 2021;12(5):514-523.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


