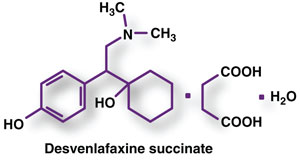
US Pharm. 2008;33(10):30-44.
New molecular entities
(NMEs), as defined by the FDA, are new drug products containing, as their
active ingredient, a chemical substance marketed for the first time in the
United States. The following descriptions of NMEs approved in 2007-2008 (TABLE)
include a brief summary of the clinical and pharmacologic profile for each new
drug, as well as selected pharmacokinetics, adverse reactions, drug
interactions, and dosing information. This review is intended to be objective
rather than evaluative in content. The information for each NME was obtained
primarily from sources published prior to FDA approval. Experience has shown
that many aspects of a new drug's therapeutic profile, such as adverse
reactions, do not emerge until after the drug is used in large numbers of
patients for several years. Hence, while this review offers a starting point
for learning about new drugs, it is essential that practitioners become aware
of changes in a drug's therapeutic profile over time.
Desvenlafaxine (Pristiq,
Wyeth Pharmaceuticals)
Indication
and Clinical Profile1-3:
Desvenlafaxine was approved in February 2008 for the treatment of major
depressive disorder (MDD). This drug is the primary active metabolite of
venlafaxine, a selective serotonin-norepinephrine reuptake inhibitor (SNRI)
that is used to treat major depressive, social anxiety, generalized anxiety,
and panic disorders. MDD affects about 121 million people worldwide, including
approximately 15 million adults in the U.S. or 6.7% of the population aged 18
and older.
The efficacy of desvenlafaxine
was assessed in four 8-week, randomized, double-blind, placebo-controlled,
fixed-dose trials involving adult patients who met the Diagnostic and
Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria
for MDD. In these trials, doses of 50 to 400 mg were shown to produce greater
improvement in the 17-item Hamilton Rating Scale for Depression (HAM-D17)
total score compared with placebo. In three of the four studies,
desvenlafaxine was also associated with greater overall improvement, assessed
using the Clinical Global Impressions Scale-Improvement (CGI-I) versus placebo.
Pharmacology and
Pharmacokinetics1-3:
Desvenlafaxine, like venlafaxine, is a potent, selective SNRI. Its clinical
efficacy is believed to be related to the potentiation of these
neurotransmitters in the central nervous system (CNS). Desvenlafaxine does not
display significant affinity for numerous other receptors, including
muscarinic-cholinergic, H1-histaminergic, or alpha1-adrenergic
receptors in vitro. It also lacks monoamine oxidase inhibitory activity.
The absolute oral
bioavailability of desvenlafaxine after oral administration is about 80%.
Administration with food (high-fat meal) increases the maximum concentration (Cmax)
by about 16%, but area under the curve (AUC) is not significantly altered.
Thus, desvenlafaxine can be taken without regard to meals. The plasma protein
binding is low (30%) and the volume of distribution at steady-state following
IV administration is 3.4 L/kg, indicating distribution into nonvascular
compartments. Desvenlafaxine is primarily metabolized by glucuronide
conjugation (mediated by uridine 5'-diphosphate glucuronosyltransferase [UGT]
isoforms) with a small amount of oxidative metabolism (N-demethylation)
mediated by CYP3A4. Approximately 45% of desvenlafaxine is excreted unchanged
in urine at 72 hours after oral administration, with 19% excreted as the
glucuronide metabolite and less than 5% as the oxidative metabolite (N,
O-didesmethylvenlafaxine). Pharmacokinetic analyses showed that gender, race,
and hepatic function have no significant apparent effect on the
pharmacokinetics of desvenlafaxine; thus, no dosage adjustment is needed.
However, due to significant increases in AUC with declining renal function,
dosage adjustment is recommended in patients with significant renal impairment.
Adverse Reactions1-3:
The most commonly observed adverse reactions in patients taking desvenlafaxine
for MDD in short-term, fixed-dose studies (incidence >=5% and at least
twice the rate of placebo in the 50- or 100-mg dose groups) were nausea,
dizziness, insomnia, hyperhidrosis, constipation, somnolence, decreased
appetite, anxiety, and specific male sexual function disorders. The drug
carries a boxed warning concerning the increased risk of suicidal thinking and
behavior in patients taking antidepressants for MDD; thus, patients should be
monitored. Activation of mania/hypomania also has occurred in a small
percentage of patients with MDD treated with desvenlafaxine. Desvenlafaxine
therapy has been associated with serotonin syndrome, increased risk of
bleeding, mydriasis, interstitial lung disease, eosinophilic pneumonia, and
elevated blood pressure, cholesterol, and triglyceride levels. The agent may
exacerbate cardiovascular/cerebrovascular disease as well as seizure
disorders. In addition, patients should notify their physician if they become
pregnant or are breastfeeding during desvenlafaxine therapy (Pregnancy
Category C).
Drug Interactions1-3:
The risk of using desvenlafaxine in combination with other CNS-active drugs
has not been systematically evaluated. Consequently, caution is advised when
desvenlafaxine is taken in combination with other CNS-active drugs. A clinical
study has shown that desvenlafaxine does not increase the impairment of mental
and motor skills caused by ethanol. However, as with all CNS-active drugs,
patients should be advised to avoid alcohol consumption while taking
desvenlafaxine. In addition, adverse reactions, some of which were serious,
have been reported in patients who have recently been discontinued from a
monoamine oxidase inhibitor (MAOI) and started on antidepressants with
pharmacologic properties similar to desvenlafaxine's (i.e., SNRIs or selective
serotonin reuptake inhibitors [SSRIs]), or who have recently had SNRI or SSRI
therapy discontinued prior to initiation of an MAOI.
Based on the mechanism of
action of desvenlafaxine and the potential for serotonin syndrome, caution is
advised when desvenlafaxine is coadministered with other drugs that may affect
the serotonergic neurotransmitter system. A number of studies have
demonstrated an association between use of psychotropic drugs that interfere
with serotonin reuptake and the occurrence of upper gastrointestinal bleeding.
These studies have also shown that concurrent use of a nonsteroidal
anti-inflammatory drug (NSAID) or aspirin may potentiate this risk of
bleeding. Altered anticoagulant effects have been reported when SSRIs and
SNRIs are coadministered with warfarin, and thus patients receiving warfarin
therapy should be carefully monitored when desvenlafaxine is initiated or
discontinued.
Based on in vitrodata, drugs
that inhibit CYP isozymes 1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19, and 2E1 are not
expected to have significant impact on the pharmacokinetic profile of
desvenlafaxine. Even though CYP3A4 is a minor pathway for the metabolism of
desvenlafaxine, potent inhibitors of CYP3A4 such as ketoconazole may result in
higher desvenlafaxine concentrations (increased AUC). Caution is advised when
using desvenlafaxine with potent inhibitors of CYP3A4. In vitro studies
showed a weak inhibitory effect of desvenlafaxine on CYP2D6. Thus, concomitant
use of desvenlafaxine with a drug metabolized by CYP2D6 such as desipramine
can result in higher concentrations of that drug.
In vitro, desvenlafaxine does not
inhibit CYP1A2, 2A6, 2C8, 2C9, and 2C19 isozymes and is not a substrate or an
inhibitor for the P-glycoprotein (P-gp). As a result, it would not be expected
to affect the pharmacokinetics of drugs that are metabolized by these CYP
isozymes or transported by P-gp. While desvenlafaxine does not inhibit or
induce CYP3A4, it can compete with other drugs metabolized by this isozyme
(e.g., midazolam) and increase their plasma concentrations.
Dosage and Administration1-3:
Desvenlafaxine is supplied as 50- and 100-mg extended-release tablets. The
recommended dosage is 50 mg once daily with or without food. When therapy is
discontinued, the dosage should be gradually tapered to minimize
discontinuation symptoms. The recommended dosage for patients with moderate
renal impairment (CrCl = 30-50 mL/min) is 50 mg/day. Patients with severe
renal impairment (CrCl <30 mL/min) or end-stage renal disease should be dosed
at 50 mg every other day.
Ambrisentan (Letairis,
Gilead Sciences)
Indication
and Clinical Profile4-6:
Ambrisentan is specifically indicated for the treatment of pulmonary arterial
hypertension (PAH) in subjects with World Health Organization (WHO) class II
or III symptoms to improve exercise capacity and delay clinical worsening.
This agent was granted orphan drug status by the FDA because only about
100,000 Americans have PAH. PAH is caused by narrowing and clot formation in
the small arteries of the lungs, resulting in continuously high pulmonary
arterial blood pressure (>=25 mmHg). This results in an increase in heart
workload and, over time, a weakening of heart muscle and reduced blood supply
to the lungs. Symptoms of PAH include shortness of breath, fatigue, chest
pain, dizzy spells, and fainting. While there is no cure for PAH, drug
therapies include anticoagulants, calcium channel blockers, nitric oxide,
sildenafil, diuretics, prostacyclins (e.g., epoprostenol, treprostinil,
iloprost), and endothelin antagonists. These therapies provide some benefit by
reducing clot formation, relaxing pulmonary arteries, and improving blood
supply and heart performance.
The efficacy and safety of
ambrisentan was evaluated in two 12-week, randomized, placebo-controlled,
multicenter studies (ARIES-1 and ARIES-2) involving 393 patients who had
idiopathic PAH or PAH associated with other disease or anorexigen drug use. In
ARIES-1, patients were treated with 5 or 10 mg once-daily doses of ambrisentan
or placebo, while in ARIES-2, patients received 2.5- or 5-mg once-daily doses
of ambrisentan or placebo. In both studies, ambrisentan or placebo was added
to a patient's current PAH therapy, which could include drugs previously
mentioned. The primary end point was six-minute walking distance (6MWD). After
12 weeks in the ARIES-1 study, ambrisentan patients demonstrated a
statistically significant improvement in 6MWD compared with patients treated
with placebo. Similar results were obtained in the ARIES-2 study. In both
studies, an increase in 6MWD was observed after four weeks of ambrisentan
treatment, and a dose-response was observed after 12 weeks of treatment. In
both studies, ambrisentan-treated patients also experienced a significant
delay in the time to clinical worsening compared with placebo. In addition,
ambrisentan was evaluated in an open-label, long-term, follow-up trial
involving 383 patients who had been previously treated in ARIES-1 and ARIES-2.
Results showed that 95% were still alive after one year and 94% continued to
receive ambrisentan monotherapy.
Pharmacology and
Pharmacokinetics4-6:
Ambrisentan is an endothelin receptor antagonist. Endothelin is an endogenous
peptide synthesized in the endothelium, and plasma endothelin concentrations
may be increased as much as 10-fold in patients with PAH. There are two
classes of endothelin receptors: endothelin type A (ETA) and
endothelin type B (ETB). The binding of endothelin to ETA
receptors results in vasoconstriction, while binding to ETB causes
vasodilation. Both bosentan and ambrisentan function as ETA-receptor
antagonists and thereby prevent the constriction or narrowing of blood vessels
and enhance blood flow throughout the body. Ambrisentan reportedly differs
from bosentan in that it has a higher degree of selectivity for the desired ETA
target receptor versus the ETB receptor.
Ambrisentan is rapidly
absorbed, providing peak concentrations in approximately two hours. While the
absolute bioavailability is not known, food does not appear to affect its
bioavailability. Ambrisentan is highly bound to plasma proteins (99%).
Elimination is predominantly by nonrenal pathways, but the relative
contributions of metabolism and biliary elimination have not been well
characterized. Based on in vitro data, metabolism may occur by CYP3A4,
CYP2C19, and UGTs. Although ambrisentan has a 15-hour terminal half-life, the
mean trough concentration at steady-state is about 15% of the mean peak
concentration and the accumulation factor is about 1.2 after long-term daily
dosing, indicating that the effective half-life is about nine hours.
Ambrisentan is not recommended in patients with moderate or severe hepatic
impairment and should be used with caution in patients with mild hepatic
impairment.
Adverse Reactions4-6:
The most common adverse events reported in patients treated with ambrisentan
include peripheral edema (swelling of legs and ankles), nasal congestion,
sinusitis, flushing, palpitations, nasopharyngitis, abdominal pain,
constipation, dyspnea, and headache. Treatment with ET-receptor antagonists
also has been associated with dose-dependent hepatic injury, manifested
primarily by serum aminotransferase (ALT and AST) elevations but sometimes
accompanied by abnormal liver function (i.e., bilirubin elevations). If
aminotransferase elevations are accompanied by clinical symptoms of liver
injury (e.g., nausea, vomiting, fever, abdominal pain, jaundice, unusual
lethargy, or fatigue) or increases in bilirubin greater than two times the
upper limit of normal (ULN), treatment should be stopped. Patients treated
with ET-receptor antagonists have also experienced decreases in hemoglobin
concentration and hematocrit, and thus these parameters should be monitored.
Ambrisentan may cause fetal harm if administered to a pregnant woman, so this
agent is contraindicated in women who are or who may become pregnant
(Pregnancy Category X). Because of the risks of liver injury and birth
defects, ambrisentan is available only through a special restricted
distribution program called the Letairis Education and Access Program (LEAP).
Drug Interactions4-6:
Ambrisentan is metabolized by CYP3A4, CYP2C19, and the UGTs 1A9S, 2B7S, and
1A3S. It is also a substrate, but not an inhibitor, of the organic anion
transport protein (OATP) and P-gp transporters. The drug interaction potential
of ambrisentan with strong inducers or inhibitors of CYP3A4 or CYP2C19, or
strong inhibitors of the transporters P-gp (i.e., cyclosporine A) and OATP
(i.e., cyclosporine A, rifampin), has not been characterized. Thus, the impact
of coadministration of such drugs on ambrisentan exposure is unknown, and
caution is advised. Dosage adjustment does not appear to be required when
ambrisentan is used with other drugs to treat PAH, including warfarin and
sildenafil.
Dosage and Administration4-6:
Ambrisentan is supplied as 5- or 10-mg film-coated, unscored
tablets designed for oral administration. These tablets may be administered
with or without food, but should not be split, crushed, or chewed. The
recommended initial dosage of the drug is 5 mg once daily. The dosage may be
increased to 10 mg once daily, but doses higher than 10 mg once daily have not
been studied. Liver function tests should be measured prior to initiation and
during treatment, and the drug should not be used in patients with moderate or
severe hepatic impairment.
Raltegravir (Isentress,
Merck & Co., Inc.)
Indication
and Clinical Profile7-9:
Raltegravir, in combination with other antiretroviral therapy,
is approved for the treatment of HIV-1 infection in treatment-experienced
patients with ongoing viral replication despite existing therapy. The drug is
the first in a new class of antiretroviral agents called integrase
inhibitors. Raltegravir received "priority review status," a designation
for investigational products that address unmet medical needs.
The efficacy of raltegravir
was assessed in two randomized, double-blind, placebo-controlled trials
(BENCHMRK 1 and BENCHMRK 2). These trials enrolled antiretroviral
treatment-experienced adult patients (aged >=16 years) with HIV-1
infection resistant to one or more drugs in each of three classes of
antiretroviral therapies (nucleoside reverse transcriptase inhibitors [NRTIs],
nonnucleoside reverse transcriptase inhibitors [NNRTIs], or protease
inhibitors [PIs]). Patients were randomized to receive raltegravir 400 mg
twice daily plus optimized background therapy (OBT) or placebo plus OBT. After
24 weeks of treatment, 75.5% of patients who received raltegravir plus OBT
demonstrated HIV-1 RNA less than 400 copies/mL versus 39.3% of patients
treated with placebo plus OBT. Additionally, 62.6% of the raltegravir-treated
patients demonstrated HIV-1 RNA less than 50 copies/mL versus 33.3% of
patients who received placebo plus OBT. Patients treated with raltegravir plus
OBT demonstrated a mean change from baseline in plasma HIV-1 RNA of -1.85 log10
copies/mL versus -0.84 log10 copies/mL in patients treated with
placebo plus OBT. Raltegravir-treated patients demonstrated a mean increase
from baseline in CD4+ cell counts of 89 cells/mm3 versus 35 cells/mm3
among patients treated with placebo plus OBT.
Pharmacology and
Pharmacokinetics7-9:
Raltegravir is an HIV integrase strand transfer inhibitor. By inhibiting the
catalytic activity of this enzyme, raltegravir prevents the covalent
integration of unintegrated linear HIV-1 DNA into the host cell genome, thus
preventing the formation of the HIV-1 provirus and propagation of the viral
infection. Raltegravir does not significantly inhibit human
phosphoryltransferases including DNA polymerases alpha, beta, and gamma.
Additive to synergistic antiretroviral activity was observed when certain
HIV-infected human cell lines were incubated with raltegravir in combination
with NNRTIs, NRTIs, PIs, or the entry inhibitor enfuvirtide. The mutations
observed in the HIV-1 integrase coding sequence that contribute to raltegravir
resistance (evolved either in cell culture or in subjects treated with
raltegravir) generally include an amino acid substitution at either Q148
(changed to H, K, or R) or N155 (changed to H), plus one or more additional
substitutions (i.e., L74M/R, E92Q, T97A, E138A/K, G140A/S, V151I, G163R,
H183P, Y226D/F/H, S230R, and D232N). Amino acid substitution at Y143C/H/R is
another pathway to raltegravir resistance.
Raltegravir is readily
absorbed upon oral administration, producing peak plasma levels within three
hours. The absolute bioavailability of raltegravir has not been determined,
but absorption appears to be increased when taken with food. Raltegravir is
approximately 83% bound to human plasma protein. The major pathway of
raltegravir metabolism is glucuronidation mediated by the UGT1A1 isoform of
the enzyme. The parent drug is the primary circulating drug entity (70%),
while the glucuronide accounts for the minor circulating species. The apparent
terminal half-life is approximately nine hours, with a shorter alpha-phase
half-life (~1 hour) accounting for much of the AUC. Approximately 51% and 32%
of the oral dose is excreted in feces and urine, respectively. In feces, only
raltegravir is present, most of which is likely derived from hydrolysis of
raltegravir-glucuronide excreted in bile. Both raltegravir and its glucuronide
are excreted in urine, accounting for approximately 9% and 23% of the dose,
respectively. The effect of severe hepatic impairment on the pharmacokinetics
of raltegravir has not been studied.
Adverse Reactions7-9:
The most common adverse events observed in raltegravir-treated patients in
clinical trials included diarrhea, nausea, headache, and pyrexia. When
treatment is initiated, some patients developed immune reconstitution
syndrome, an inflammatory response to indolent or residual opportunistic
infections. More rare, but serious, drug-related reactions reported with
raltegravir in clinical trials included hypersensitivity, anemia, neutropenia,
gastritis, myocardial infarction, hepatitis, herpes simplex, toxic
nephropathy, renal failure, chronic renal failure, and renal tubular necrosis.
Rhabdomyolysis and myopathy were also reported, but the possible relationship
between these events and raltegravir treatment is unknown. Raltegravir is a
Pregnancy Category C drug and should be used in pregnancy only if the
potential benefit justifies the potential risk to the fetus.
Drug Interactions7-9:
Raltegravir is not a substrate, inducer, or inhibitor of the cytochrome
isozymes CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A. Similarly,
raltegravir is not an inhibitor of the UGT isozymes UGT1A1 and UGT2B7, and it
also does not inhibit P-gp–mediated transport. Based on these data,
raltegravir is not expected to affect the pharmacokinetics of drugs that are
substrates of these enzymes or P-gp.
Since raltegravir is a
substrate for UGT1A1, strong inducers of this enzyme, such as rifampin, can
significantly reduce raltegravir plasma concentrations. Thus, caution should
be used when coadministering raltegravir with strong inducers of UGT1A1. The
impact of other inducers of drug-metabolizing enzymes (e.g., phenytoin and
phenobarbital) on UGT1A1 is unknown. Other less strong inducers (e.g.,
efavirenz, nevirapine, rifabutin, St. John's wort) may be used with the
recommended dose of raltegravir.
Dosage and Administration7-9:
Raltegravir is supplied as 400-mg film-coated tablets. It should be
administered at a dosage of 400 mg twice daily with or without food. No
dosage adjustments are necessary based on age, gender, race, or renal
impairment or in mild to moderate hepatic impairment.
Etravirine (Intelence,
Tibotec Therapeutics)
Indication
and Clinical Profile10-12:
Etravirine, in combination with other antiretroviral agents, is indicated for
the treatment of HIV-1 infection in antiretroviral treatment-experienced adult
patients who have evidence of viral replication and HIV-1 strains resistant to
an NNRTI and other antiretroviral agents. This drug is the first new NNRTI to
be introduced in nearly 10 years.
Approval of etravirine was
based on pooled 24-week results of two ongoing, randomized, placebo-controlled
Phase III trials designated as DUET-1 and DUET-2. The trials were designed to
evaluate the safety and antiretroviral activity of etravirine in combination
with a background regimen (BR), as compared to placebo in combination with a
BR. Randomization was stratified by the intended use of enfuvirtide (ENF) in
the BR, previous use of darunavir/ritonavir (DRV/rtv), and screening viral
load. All study subjects received DRV/rtv as part of their BR, and at least
two other investigator-selected antiretroviral drugs (nucleotide reverse
transcriptase inhibitors [NtRTIs] with or without ENF). At week 24, 74% of
etravirine-treated subjects achieved HIV-1 RNA less than 400 copies/mL as
compared to 51.5% of placebo-treated subjects. The mean decrease in plasma
HIV-1 RNA from baseline to week 24 was -2.37 log10 copies/mL for
etravirine-treated subjects and -1.68 log10 copies/mL for
placebo-treated subjects. The mean CD4+ cell count increase from baseline was
81 cells/mm3 for etravirine-treated subjects and 64 cells/mm3
for placebo-treated subjects. Of the population who either reused or did not
use ENF, 56.7% of etravirine-treated subjects and 32.7% of placebo-treated
subjects achieved HIV-1 RNA less than 50 copies/mL, the end point virologic
response. Of the study population using ENF for the first time, 68.6% of
etravirine-treated subjects and 61.3% of placebo-treated subjects achieved
HIV-1 RNA less than 50 copies/mL.
Pharmacology and
Pharmacokinetics10-12:
Etravirine is an NNRTI of HIV-1. Reverse transcriptase is a viral DNA
polymerase enzyme that HIV needs to replicate. Etravirine binds directly to
reverse transcriptase and blocks the RNA-dependent and DNA-dependent DNA
polymerase activities by causing a disruption of the enzyme's catalytic site.
This prevents completion of synthesis of the double-stranded viral DNA, thus
blocking HIV replication. Etravirine did not show antagonism when studied in
combination with other NNRTIs, NtRTIs, PIs, or the fusion inhibitor ENF.
Etravirine also does not inhibit the human DNA polymerases alpha, beta, and
gamma.
In clinical trials, virologic
failure or resistance to an etravirine-containing regimen was observed most
commonly in those patients infected with HIV-1 strains with reverse
transcriptase substitutions (mutations) at positions V179F, V179I, Y181C, and
Y181I. These usually emerged in a background of multiple other NNRTI
resistance-associated substitutions. Cross-resistance to the other NNRTIs
delavirdine, efavirenz, and/or nevir apine is expected after virologic
failure with an etravirine-containing regimen.
Etravirine is readily absorbed
following oral administration, producing peak plasma levels within four hours.
The absolute oral bioavailability of etravirine is unknown; however, it should
always be taken with food to optimize absorption. Absorption is not affected
by coadministration with drugs that increase gastric pH, including ranitidine
or omeprazole. Etravirine is about 99.9% bound to plasma proteins, primarily
to albumin (99.6%) and alpha1-acid glycoprotein (97.66%-99.02%).
Distribution into compartments other than plasma (e.g., cerebrospinal fluid,
genital tract secretions) has not been evaluated in humans.
Etravirine primarily undergoes
metabolism by CYP3A4, CYP2C9, and CYP2C19 enzymes. The major metabolites,
formed by methyl hydroxylation of the dimethylbenzonitrile moiety, are less
active than the parent drug. The majority of the oral dose (94%) is eliminated
in the feces, with unchanged drug accounting for the majority (>80%) of the
excretion product. The mean terminal elimination half-life of etravirine is
about 40 hours. No significant pharmacokinetic differences have been observed
based on age, gender, race, or mild to moderate hepatic impairment. The
pharmacokinetics of etravirine has not been studied in patients with severe
hepatic impairment or renal impairment.
Adverse Reactions10-12:
In clinical trials, the most common treatment-emergent adverse reactions
(Grade 2-4) that occurred in 2% or more of patients receiving an
etravirine-containing regimen were diarrhea, nausea, abdominal pain, vomiting,
fatigue, peripheral neuropathy, headache, rash, and hypertension. The rashes
were mild to moderate, occurred primarily in the second week of therapy, and
generally resolved within one to two weeks on continued therapy. Severe and
potentially life-threatening skin reactions, including Stevens-Johnson
syndrome, have been reported (<0.1%) in patients taking etravirine. Treatment
with etravirine should be discontinued and appropriate therapy initiated if
severe rash develops. In general, immune reconstitution syndrome and
redistribution and/or accumulation of body fat have been observed in patients
receiving antiretroviral therapy. Etravirine can be used during pregnancy if
the potential benefit justifies the potential risk (Pregnancy Category B).
Mothers should not breastfeed due to the potential for HIV transmission.
Drug Interactions10-12:
Etravirine is a substrate of the isozymes CYP3A4, CYP2C9, and CYP2C19.
Therefore, coadministration of etravirine with drugs that induce or inhibit
CYP3A4, CYP2C9, and CYP2C19 may alter the therapeutic effect or the adverse
reaction profile of the coadministered drug. Etravirine is also an inducer of
CYP3A4 and inhibitor of CYP2C9 and CYP2C19. An extensive listing of drugs with
established or other potentially significant drug interactions based on which
alterations in dose or regimen of etravirine and/or a coadministered drug are
provided in the manufacturer's literature.
Dosage and Administration10-12:
Etravirine is supplied as a 100-mg tablet designed for oral administration.
The recommended initial dosage of the drug is 200 mg (two 100-mg tablets)
taken twice daily following a meal. If a patient is unable to swallow a tablet
whole, it may be dispersed in a glass of water and drunk immediately.
Maraviroc (Selzentry,
Pfizer)
Indication
and Clinical Profile13-15:
Maraviroc is approved for use in combination with other antiretroviral drugs
for the treatment of adults with CCR5-tropic HIV-1 (also known as the R5
virus) who have been treated with other HIV medications and who have
evidence of elevated levels of HIV in their blood (viral load). CCR5 is a
protein on the surface of some types of immune cells, and its receptor
component, the CCR5 coreceptor, is the predominant route of entry of HIV virus
into these cells. Maraviroc prevents the virus from entering uninfected cells
by blocking the CCR5 co-receptor. Among patients who have previously received
HIV medications, approximately 50% to 60% have circulating CCR5-tropic HIV-1.
The efficacy of maraviroc was
evaluated by analyses of 24-week data from two ongoing multicenter studies
(MOTIVATE-1 and MOTIVATE-2), designated as enrolling adult patients with
CCR5-tropic HIV-1 and with HIV-1 RNA greater than 5,000 copies/mL in spite of
six months or more of prior therapy with one or more antiretroviral agents
from three of the four antiretroviral drug classes or with documented
resistance or intolerance to one or more member of each class. All patients
were treated with an optimized BR of three to six antiretroviral agents
(excluding low-dose ritonavir) based on the patient's treatment history and
baseline genotypic and phenotypic viral resistance measurements. Patients were
randomized 2:2:1 to maraviroc 300 mg once daily, maraviroc 300 mg twice daily,
or placebo. Doses were adjusted based on background therapy. After 24 weeks of
therapy, 60.8% of patients treated with maraviroc 300 mg twice daily had HIV-1
RNA less than 400 copies/mL versus 27.8% of patients who received placebo.
From baseline to week 24, patients treated with maraviroc 300 mg twice daily
experienced a mean change in HIV-1 RNA of -1.96 log10.
In Study A4001029, patients
were required to meet inclusion/exclusion criteria similar to those of
MOTIVATE-1 and MOTIVATE-2. Patients were randomized 1:1:1 to maraviroc once
daily, maraviroc twice daily, or placebo. Patients treated with maraviroc
demonstrated no increased risk of infection or HIV disease progression.
Maraviroc use among these patients was not associated with a significant
decrease in HIV-1 RNA versus placebo-treated patients.
Pharmacology and
Pharmacokinetics13-15:
Maraviroc is an HIV-1 entry inhibitor that works by blocking the virus from
entering human cells. Specifically, maraviroc is a selective, slowly
reversible, small-molecule antagonist of the interaction between human CCR5
and HIV-1 gp120. CXCR4-tropic and dual-tropic HIV-1 entry is not inhibited by
maraviroc. While the resistance profile in treatment-naïve and
treatment-experienced subjects has not been fully characterized, virologic
failure can result from genotypic and phenotypic resistance to maraviroc or
through outgrowth of undetected CXCR4-using virus present before maraviroc
treatment. In treatment-experienced patients, resistant viruses have emerged
with multiple amino acid substitutions with unique patterns in the
heterogeneous V3 loop region of gp120.
The absolute bioavailability
of maraviroc over the therapeutic dose range is 23% to 33%, and peak plasma
concentrations are reached in 0.5 to four hours. Maraviroc is bound (~76%) to
human plasma proteins and shows moderate affinity for albumin and alpha1-acid
glycoprotein. The volume of distribution of maraviroc is approximately 194 L.
In vitro studies indicate that CYP3A is the primary enzyme responsible for
maraviroc metabolism. The parent drug (~42%) and N-dealkyl metabolite
(~22%) are the predominant circulating species in plasma. Other metabolites
are formed from CYP-based mono-oxidation and are only minor components in
plasma, possessing essentially no antiretroviral activity. Maraviroc is a
substrate for the efflux transporter P-gp. The terminal half-life of maraviroc
following oral dosing to steady state is 14 to 18 hours. Approximately 20% of
the oral dose is eliminated in the urine and 76% in the feces over 168 hours.
Maraviroc is the major component present in urine (mean of 8% dose) and feces
(mean of 25% dose), and the remainder is excreted as metabolites.
Adverse Reactions13-15:
The most common adverse events reported in trial patients treated with
maraviroc included upper respiratory tract infections, cough, pyrexia, rash,
musculoskeletal symptoms, abdominal pain, fever, and dizziness. Maraviroc
therapy has been associated with hepatotoxicity and an increase in hepatic
adverse events. The drug should be used with caution in patients at increased
risk for cardiovascular events. The product label includes a boxed warning
about hepatotoxicity and a statement warning about the possibility of heart
attacks. Caution should be used when maraviroc is administered to patients
with a history of postural hypotension and in patients taking concomitant
medications that are known to lower blood pressure.
Combination antiretroviral
therapy has been associated with immune reconstitution syndrome. Patients
treated with maraviroc along with other antiretrovirals may be at increased
risk of developing infections. Maraviroc could also affect immune surveillance
and lead to an increased risk of malignancy.
Drug Interactions13-15:
Maraviroc is a substrate of CYP3A and P-gp. Its pharmacokinetics may be
altered by inhibitors and inducers of these enzymes/transporters, and dose
adjustment may be required when maraviroc is coadministered with those drugs.
CYP3A/P-gp inhibitors ketoconazole, lopinavir/ritonavir, ritonavir,
saquinavir, and atazanavir are all reported to increase the Cmax
and AUC of maraviroc. The CYP3A inducers rifampin and efavirenz decreased the Cmax
and AUC of maraviroc. Tipranavir/ritonavir (net CYP3A inhibitor/P-gp inducer)
did not affect the steady state pharmacokinetics of maraviroc. Concomitant use
of maraviroc and St. John's wort is not recommended since these products may
substantially decrease maraviroc concentrations, resulting in suboptimal
maraviroc levels, loss of virologic response, and possible resistance to
maraviroc.
Dosage and Administration13-15:
Maraviroc is supplied as 150- and 300-mg film-coated tablets. It must be given
in combination with other antiretroviral agents. The recommended initial
dosage of maraviroc differs based on concomitant medications due to drug
interactions. When given concomitantly with strong CYP3A inhibitors such as
PIs (except tipranavir/ritonavir), delavirdine, ketoconazole, itraconazole,
clarithromycin, telithromycin, nefazodone, and with or without a CYP3A
inducer, a dose of 150 mg should be administered twice daily. When given with
CYP3A inducers (i.e., efavirenz, rifampin, carbamazepine, phenobarbital,
phenytoin) without a strong CYP3A inhibitor, the maraviroc dosage is 600 mg
twice daily. When administered concurrently with medications such as
tipranavir/ritonavir, nevirapine, NRTIs, and enfuvirtide, the maraviroc dose
is 300 mg twice daily.
REFERENCES
1. Pristiq
(desvenlafaxine) package insert. Philadelphia, PA: Wyeth Pharmaceuticals Inc;
February 2008.
2. Liebowitz MR, Yeung
PP, Entsuah R. A randomized, double-blind, placebo-controlled trial of
desvenlafaxine succinate in adult outpatients with major depressive disorder. J
Clin Psychiatry. 2007;68:1663-1672.
3. Septien-Velez L,
Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind,
placebo-controlled trial of desvenlafaxine succinate in the treatment of major
depressive disorder. Int Clin Psychopharmacol. 2007;22:338-347.
4. Letairis
(ambrisentan) package insert. Foster City, CA: Gilead Sciences, Inc; February
2008.
5. Vatter H, Seifert V.
Ambrisentan, a non-peptide endothelin receptor antagonist. Cardiovas Drug
Rev. 2006;24:63-76.
6. Galié N, Badesch D,
Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J
Am Coll Cardiol. 2005;46:529-535.
7. Isentress
(raltegravir) package insert. Whitehouse Station, NJ: Merck & Co., Inc;
October 2007.
8. Markowitz M, Nguyen
BY, Gotuzzo E, et al; Protocol 004 Part II Study Team. Rapid and durable
antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of
combination therapy in treatment-naive patients with HIV-1 infection: results
of a 48-week controlled study. J Acquir Immune Defic Syndr.
2007;46:125-133.
9. Grinsztejn B, Nguyen
BY, Katlama C, et al; Protocol 005 Team. Safety and efficacy of the HIV-1
integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients
with multidrug-resistant virus: a phase II randomised controlled trial. Lancet.
2007;369:1261-1269.
10. Intelence
(etravirine) package insert. Raritan, NJ: Tibotec Therapeutics; January 2008.
11. Lazzarin A,
Campbell T, Clotet B, et al; DUET-2 study group. Efficacy and safety of TMC125
(etravirine) in treatment-experienced HIV-1-infected patients in DUET-2:
24-week results from a randomised, double-blind, placebo-controlled trial. Lancet.
2007;370:39-48.
12. Madruga JV, Cahn P,
Grinsztejn B, et al; DUET-1 study group. Efficacy and safety of TMC125
(etravirine) in treatment-experienced HIV-1-infected patients in DUET-1:
24-week results from a randomized, double-blind, placebo-controlled trial. Lancet.
2007;370:29-38.
13. Selzentry
(maraviroc) package insert. New York, NY: Pfizer; August 2007.
14. Fätkenheuer G,
Pozniak AL, Johnson MA, et al. Efficacy of short-term monotherapy with
maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med.
2005;11:1170-1172.
15. Rosario MC, Poland
B, Sullivan J, et al. A pharmacokinetic-pharmacodynamic model to optimize the
phase IIa development program of maraviroc. J Acquir Immune Defic Syndr.
2006;42:183-191.
To comment on this article, contact
rdavidson@jobson.com.



