US Pharm. 2010;35(10):26-51.
New molecular entities (NMEs), as defined by the FDA, are drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2009–2010 (TABLE 1) detail the basic clinical and pharmacologic profiles for each new drug, as well as its pharmacokinetics, adverse reactions, drug interactions, and dosing data. Note that the information for each NME was obtained primarily from sources published prior to FDA approval; thus, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature, such as the emergence of additional adverse reactions and black box warnings.
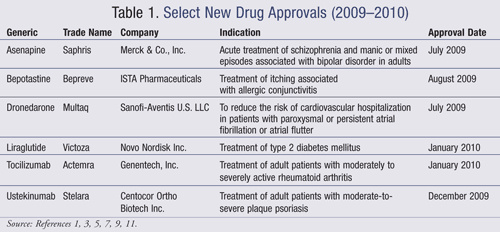
Asenapine (Saphris, Merck & Co., Inc.)
Indication and Clinical Profile1,2: Asenapine, a second-generation antipsychotic, has been approved in a sublingual tablet for acute treatment of schizophrenia and manic or mixed episodes associated with bipolar disorder. Its efficacy in schizophrenia was demonstrated in three 6-week, placebo-controlled trials with active comparator controls in patients with acute schizophrenia. In two of these trials, asenapine 5 mg twice daily was more effective than placebo in reducing symptoms of schizophrenia. In another, haloperidol 4 mg twice daily was superior to placebo, while asenapine 10 mg twice daily was not. In the second trial, 3 mg of risperidone twice daily was the active control and was not significantly more effective than placebo. In the third trial, olanzapine 15 mg daily was significantly more effective than placebo, but asenapine 5 mg twice daily was not.
The efficacy of asenapine in bipolar disorder was established in two 3-week studies of patients with acute bipolar mania or mixed states that found asenapine 10 mg twice daily, with an optional dose reduction to 5 mg twice daily, more efficacious than placebo. In a 9-week trial extension, asenapine was found to be noninferior to olanzapine in reducing the severity of mania.
Pharmacology and Pharmacokinetics1: Asenapine is a dibenzo-oxepino pyrrole, and like most second-generation antipsychotics, is an antagonist at dopamine (D2) and serotonin (5-HT2A) receptors (FIGURE 1). It has high affinity for other dopamine and serotonin receptors, as well as alpha-adrenergic and histaminergic (H1) receptors, but low affinity for muscarinic receptors. When administered sublingually, the drug is 35% bioavailable; oral bioavailability of oral asenapine is very low (<2%) due to extensive first-pass metabolism. Steady state is achieved within 3 days of twice-daily dosing. Asenapine is cleared by direct conjugation and CYP450 1A2 oxidation and is eliminated in both the urine and feces primarily as metabolites. It has a half-life of 24 hours.
Adverse Reactions1,2: The most common adverse effects, occurring in >5% of trial patients taking asenapine and at least twice as frequently as with placebo, were akathisia, oral hypoesthesia, and somnolence in schizophrenia trials, and dizziness and extrapyramidal symptoms other than akathisia in bipolar trials. Weight gain in clinical trials was greater than with placebo, but less than with risperidone or olanzapine. In short-term schizophrenia trials, serum prolactin levels increased to >4 times the upper limit of normal (ULN) in 2.6% of patients treated with asenapine, compared to 0.6% of those treated with placebo. Asenapine increases the corrected QT interval (QTc) by 2 to 5 msec and thus should not be used with other drugs that prolong the QT interval or in patients with risk factors for QT prolongation. Like other antipsychotic drugs, asenapine should not be used in elderly patients with dementia-related psychosis since these patients are at an increased risk of death (black box warning). Asenapine is a Pregnancy Category C drug and should be avoided during nursing.
Drug Interactions1: Asenapine plasma levels may increase when coadministered with strong inhibitors of CYP1A2 such as fluvoxamine. Asenapine is a weak inhibitor of CYP2D6; serum concentrations of paroxetine—a substrate and inhibitor of CYP2D6—increased almost two-fold when taken with asenapine. The risks of using asen-apine in combination with other drugs have not been extensively evaluated, but caution is advised when it is taken in combination with other centrally acting drugs or alcohol. Because of its alpha1-adrenergic antagonism with potential for inducing hypotension, asenapine may enhance the effects of certain antihypertensive agents.
Dosage and Administration1: Asenapine is supplied as 5 and 10 mg white sublingual tablets. The recommended dosage for schizophrenia is 5 mg twice daily; for bipolar disorder the recommended dosage is 10 mg twice daily, which can be reduced to 5 mg twice daily if the higher dose is poorly tolerated. Patients should not eat or drink for 10 minutes after taking the drug. It also should not be used in patients with severe hepatic impairment. Whether schizophrenic or acutely manic patients will be able to adhere to the sublingual dosing requirement is a concern.
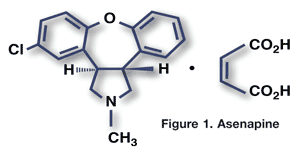
Bepotastine (Bepreve, ISTA Pharmaceuticals)
Indication and Clinical Profile3,4: Bepotastine besilate 1.5% ophthalmic solution is a H1-receptor antagonist agent approved for the topical treatment of itching associated with allergic conjunctivitis in patients 2 years of age and older. Allergic conjunctivitis is the most common form of ocular allergy, with itching as the main symptom. Treatment of ocular itching involves antihistamines (both oral and ophthalmic), ophthalmic formulations of mast cell stabilizers, nonsteroidal anti-inflammatory drugs (NSAIDs), and, in severe cases, corticosteroids. Ocular antihistamines are effective and have a more rapid onset of action than oral antihistamines. Bepotastine was first developed in Japan as an oral systemic formulation for the treatment of allergic rhinitis.
Two randomized, double-masked, placebo-controlled conjunctival allergen challenge studies conducted in 237 patients with a history of allergic conjunctivitis found bepotastine 1.5% ophthalmic solution more effective than placebo in relieving ocular itching after 15 minutes and at 8 hours.
There are no published studies comparing bepotastine ophthalmic solution to other topical drugs for allergic conjunctivitis. At present, there is no evidence that it offers any advantage over other ophthalmic H1 antihistamines.
Pharmacology and Pharmacokinetics3: Bepotastine is similar in structure to ethanolamine antihistamines (FIGURE 2) and is a topically active, direct H1-receptor antagonist and an inhibitor of mast-cell histamine release. Its onset of action is approximately 3 minutes and duration of effect is 8 hours. Bepotastine is only minimally absorbed from its application site. Absorbed drug is eliminated renally, predominately in the unchanged form.
Adverse Reactions and Drug Interactions3,4: In clinical trials, the most commonly reported adverse effects of bepotastine were mild taste disturbances, occurring in about 25% of patients. Other adverse effects include eye irritation, headache, and nasopharyngitis (2%-5%). During a 6-week safety study that included 127 children ≥3 years old, no serious adverse effects were detected. Bepotastine is a Pregnancy Category C drug and should be used with caution in pregnancy and nursing. Since it is applied topically, is minimally absorbed, and does not appear to interact with cytochrome isozymes (CYP3A4, CYP2C9, CYP2C19), no significant drug interactions are anticipated.
Dosage and Administration3: Bepotastine is supplied as a 1.5% ophthalmic solution of the besilate salt. One drop should be instilled into the affected eyes twice a day.
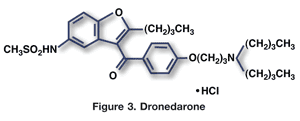
Dronedarone (Multaq, Sanofi-Aventis)
Indication and Clinical Profile5,6: Dronedarone was approved to reduce the risk of cardiovascular (CV) hospitalization in patients with paroxysmal or persistent atrial fibrillation (AF) or atrial flutter (AFL) who have experienced a recent episode of AF/AFL, have associated CV risk factors (age >70, hypertension, diabetes, previous cerebrovascular accident, left atrial diameter ≥50 mm, left ventricular ejection fraction <40%), and are in sinus rhythm or will be cardioverted. Amiodarone, the most effective drug for this indication, has a number of limitations, including toxicity.
The efficacy of dronedarone was demonstrated in several trials. Patients aged ≥75 years or ≥70 years with one or more risk factors and with a recent history of AF/AFL who were in sinus rhythm or were to be converted to sinus rhythm were randomized to treatment with dronedarone 400 mg twice/day or placebo for up to 30 months. The primary end point was time to first hospitalization for CV reasons or death from any cause. Dronedarone reduced the incidence of the primary end point by 24.2% compared with placebo; this result was entirely attributable to dronedarone’s effect on CV hospitalization. In two other trials, patients in sinus rhythm who had experienced a previous episode of AF/AFL were randomized to treatment with dronedarone 400 mg twice daily or placebo for 12 months. The primary end point was time to first recurrence of AF/AFL. Dronedarone reduced the risk of first AF/AFL recurrence by about 25%, with an approximate 11% absolute difference in recurrence rate. In another study, patients who were recently hospitalized with symptomatic heart failure (HF) and severe left ventricular systolic dysfunction were randomized to treatment with dronedarone 400 mg twice daily or placebo. This trial was terminated prematurely because of excess mortality among dronedarone-treated patients.
Pharmacology and Pharmacokinetics5,6: Dronedarone is a benzofuran, noniodinated derivative of amiodarone (FIGURE 3) with antiarrhythmic properties belonging to all four Vaughan-Williams class mechanisms. It blocks sodium, potassium, and calcium channels and has alpha- and beta-blocking properties, but which of these produce dronedarone’s clinical effects remains unclear.
Dronedarone has higher oral bioavailability (35%-65%) than amiodarone and achieves steady state levels more readily (408 days). It is also significantly less lipophilic than amiodarone, resulting in a substantially smaller volume of distribution and a much shorter elimination half-life (13-19 hours). It is extensively metabolized by CYP3A4 isozymes, initially by n-debutylation to form the active n-debutyl metabolite. This metabolite undergoes oxidative deamination to form the inactive propanoic acid metabolite and direct oxidation. These metabolites undergo further metabolism to yield over 30 uncharacterized metabolites. The n-debutyl metabolite exhibits pharmacologic activity but is significantly less potent than the parent drug. Dronedarone and its metabolites are eliminated primarily in the feces (>80%).
Adverse Reactions5,6: In trials, the most common adverse events associated with dronedarone treatment included diarrhea, nausea, abdominal pain, vomiting, and asthenia, but unlike amiodarone, significant thyroid, pulmonary, ocular, or liver toxicity has not been reported to date. Photosensitivity has been reported with dronedarone, although not the blue-gray skin discoloration associated with amiodarone. Serum creatinine levels increased by an average of 0.1 mg/dL with dronedarone treatment, but reached a plateau in 1 week and declined after discontinuation.
Dronedarone is contraindicated in patients with New York Heart Association (NYHA) class IV HF or NYHA class II or III HF with a recent decompensation requiring hospitalization or referral to a specialized HF clinic; patients with second- or third-degree atrioventricular block or sick sinus syndrome (except in patients with a functioning pacemaker); patients with bradycardia <50 beats per minute (bpm); and patients being treated with strong CYP3A inhibitors. It is also contraindicated in patients currently taking drugs that prolong the QT interval and might increase the risk of torsade de pointes (e.g., phenothiazines, tricyclic antidepressants, certain macrolide antibiotics, and class I and III antiarrhythmics) and in patients with QTc-Bazett interval ³500 msec or PR interval >280 msec. Dronedarone should not be used during pregnancy (Category X) or nursing or in patients with severe hepatic impairment.
Drug Interactions5: Based on dronedarone’s clearance profile, concomitant use of strong inhibitors of CYP3A4, such as ketoconazole, clarithromycin, or grapefruit juice, is contraindicated. Moderate inhibitors of CYP3A4 (e.g., verapamil or diltiazem) should be used with caution. Significant CYP3A4 inducers (e.g., rifampin, phenobarbital, carbamazepine, phenytoin, or St. John’s wort) could decrease the efficacy of dronedarone. Dronedarone itself is a moderate inhibitor of CYP3A4 and 2D6 and may increase serum concentrations of drugs that are substrates of these enzymes, including simvastatin, propranolol, and metoprolol. It is also a P-glycoprotein inhibitor and has increased serum concentrations of P-glycoprotein substrates, such as digoxin. Unlike amiodarone, dronedarone has not caused clinically significant increase in international normalized ratio in patients taking warfarin concurrently. It should not be used with other drugs that prolong the QT interval.
Dosage and Administration5: Dronedarone is supplied as 400-mg white, film-coated tablets. The recommended dosage is 400 mg twice daily with the morning and evening meals. No dose adjustments are recommended based on age, sex, kidney function, or moderate hepatic impairment.
Dronedarone should not be used in patients with severe hepatic impairment and class I or III antiarrhythmics. Drugs that strongly inhibit CYP3A must also be discontinued before initiation of dronedarone treatment. How to transition from amiodarone to dronedarone is unclear.
Liraglutide (Victoza, Novo Nordisk)
Indication and Clinical Profile7,8: Liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist given by subcutaneous (SC) injection, has been approved by the FDA for treatment of patients with type 2 diabetes. It can be used alone or in addition to oral antidiabetic drugs such as metformin or glimepiride. Liraglutide is not recommended for first-line therapy and is not approved for use with insulin. Liraglutide is the second GLP-1 agonist marketed in the U.S.; the first, exenatide (Byetta), has been available since 2005.
The efficacy of liraglutide was demonstrated in a number of phase III studies referred to as the Liraglutide Effect and Action in Diabetes (LEAD) trials. Four of these studies were 26 weeks in duration and one was 52 weeks. In these studies, liraglutide was evaluated as monotherapy or in combination with one or two oral hypoglycemic medications. Liraglutide was found to result in superior lowering of blood glucose than active comparators such as sulfonylureas and thiazolidinediones. Hemoglobin A1C (HbA1C) reductions for liraglutide 1.8 mg/day, as monotherapy or in combination with other oral hypoglycemics, ranged from 1.0% to 1.5% across the LEAD trials, with baseline HbA1C values ranging from 8.2% to 8.6%. Liraglutide 1.8 mg/day plus metformin reduced HbA1C by 1.0% and reduced weight by approximately 6 lb. In another comparative trial, liraglutide (1.8 mg daily) was found to be superior to exenatide (10 mcg twice daily) in reducing HbA1C (1.12% vs. 0.79%). Currently, no studies have provided conclusive evidence that liraglutide can reduce the incidence of macrovascular complications.
Pharmacology and Pharmacokinetics7: Liraglutide is an acylated human GLP-1 receptor agonist with 97% amino acid sequence homology to endogenous human GLP-1(7-37). GLP-1(7-37) represents <20% of total circulating endogenous GLP-1. Like GLP-1(7-37), liraglutide activates the GLP-1 receptor, a membrane-bound cell-surface receptor coupled to adenylyl cyclase by the stimulatory G-protein in pancreatic beta cells. This results in increased intracellular cyclic AMP (cAMP) and promotes insulin release in the presence of elevated glucose concentrations. This insulin secretion subsides as blood glucose concentrations decrease and approach euglycemia. Liraglutide also decreases glucagon secretion in a glucose-dependent manner, delays gastric emptying, and promotes satiety and weight loss.
GLP-1(7-37) has a half-life of 1.5 to 2 minutes due to rapid degradation by the ubiquitous endogenous enzymes dipeptidyl peptidase IV (DPP-IV) and neutral endopeptidases (NEP). Unlike native GLP-1, liraglutide is stable against metabolic degradation by both peptidases and has a plasma half-life of 13 hours after SC administration. The pharmacokinetic profile of liraglutide, which makes it suitable for once-daily administration, is a result of self-association that delays absorption, plasma protein binding, and stability against metabolic degradation by DPP-IV and NEP.
Adverse Reactions7,8: The most common adverse effects of liraglutide in clinical trials were nausea (sometimes severe), diarrhea, vomiting, dyspepsia, and constipation. Hypoglycemia was also reported, particularly in those patients taking a sulfonylurea with liraglutide. Fewer than 10% of trial patients developed antiliraglutide antibodies, and antibody formation was not associated with decreased drug effectiveness. Pancreatitis, which has occurred with exenatide, has also been reported with liraglutide. Renal insufficiency and acute renal failure have occurred with exenatide, especially in patients who have pre-existing kidney disease or are taking other drugs with a potential for nephrotoxicity. To date, renal toxicity has not been reported with liraglutide. In animal studies (rats), thyroid C-cell carcinomas have developed at high liraglutide doses (8 times the clinical dose), and in human trials, thyroid C-cell hyperplasia has been reported. Thus, the FDA has required a black box warning about the risk of thyroid cancer. Liraglutide is classified as Pregnancy Category C drug and should be used with caution if nursing.
Drug Interactions7: Hypoglycemia is a concern in patients taking liraglutide concurrently with an insulin secretagogue such as a sulfonylurea, so the dose of the secretagogue may need to be lowered. Liraglutide may delay gastric emptying; thus, it could decrease serum concentrations or increase the time to maximum concentration (tmax) of oral drugs taken concurrently.
Dosage and Administration7: Liraglutide is available in 3-mL prefilled pens, each containing 18 mg (6 mg/mL) of liraglutide. Each pen can deliver doses of 0.6, 1.2, or 1.8 mg. One pen can deliver a total of 15 doses of 1.2 mg or 10 doses of 1.8 mg. The drug should be injected subcutaneously in the abdomen, thigh, or upper arm at any consistent time of the day; the injection site and timing can be changed without dose adjustment. The initial dose of liraglutide is 0.6 mg once/day for 1 week to reduce gastrointestinal (GI) adverse effects; this is not considered an effective dose. The dose should then be increased to 1.2 mg/day for 1 week; if this does not lower glucose sufficiently, the dose can be increased to 1.8 mg/day.
Tocilizumab (Actemra, Genentech)
Indication and Clinical Profile9,10: Tocilizumab has been approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response to one or more tumor necrosis factor (TNF) inhibitor therapies (i.e., infliximab). RA is a chronic, progressive inflammatory disease of the synovium of the joints and surrounding tissues and is associated with intense pain, irreversible joint destruction, and systemic complications. It is estimated to affect 1.3 million Americans. Several key cytokines are involved and are elevated in the inflammatory process, including interleukin-6 (IL-6). Tocilizumab is the first IL-6 receptor-inhibiting monoclonal antibody approved to treat RA and may be used alone or in combination with methotrexate or other disease modifying antirheumatic drugs (DMARDs). It has been approved for a number of inflammatory disease states, including RA in Japan (2005) and the European Union (2009).
The efficacy of tocilizumab has been demonstrated in five multicenter controlled trials that enrolled adult patients with RA having at least eight tender and six swollen joints at baseline. Administered every 4 weeks, tocilizumab was evaluated as monotherapy and in combination with methotrexate in patients who had not responded adequately to DMARDs, or in combination with methotrexate in patients not responding to one or more TNF antagonist therapies. In each of these trials, the proportion of patients achieving the American College of Rheumatology 20, 50, and 70 end points at 24 weeks while taking tocilizumab 8 mg/kg was greater than that of control patients receiving either placebo or methotrexate. Typically, patients receiving tocilizumab 4 mg/kg had a poorer response in clinical trials than those receiving 8 mg/kg.
Pharmacology and Pharmacokinetics9,10: Tocilizumab binds specifically to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R) and has been shown to inhibit IL-6–mediated signaling through these receptors. IL-6 is a pleiotropic proinflammatory cytokine produced by a variety of cell types including T and B cells, lymphocytes, monocytes, and fibroblasts. IL-6 is involved in diverse physiologic processes such as T-cell activation, induction of immunoglobulin secretion, initiation of hepatic acute phase protein synthesis, and stimulation of hematopoietic precursor cell proliferation and differentiation. IL-6 is also produced by synovial and endothelial cells, leading to local production of IL-6 in joints affected by inflammatory processes such as RA. In clinical trials, tocilizumab doses of 4 to 8 mg/kg decreased levels of C-reactive protein, rheumatoid factor, erythrocyte sedimentation rate, and serum amyloid, and increased hemoglobin levels.
Adverse Reactions9,10: In clinical trials, common tocilizumab-associated adverse events (≥5% of trial patients) included upper respiratory tract infections (e.g., common cold, sinus infections), headache, hypertension, and increased liver enzymes. Increases in liver enzymes (aspartate [AST] or alanine aminotransferase [ALT]) generally were mild and reversible and did not result in apparent permanent or clinically evident hepatic injury. Laboratory changes included increases in total cholesterol and decreases in neutrophils and platelets.
More serious and less common side effects of tocilizumab included perforation of the GI tract, increased risk of hepatitis B infection in those already carrying the virus, increased risk of nervous system problems (multiple sclerosis), and hypersensitivity reactions including anaphylaxis. Patients treated with tocilizumab are also at an increased risk for developing serious infections that may lead to hospitalization or death, including tuberculosis and bacterial, invasive fungal, viral, or other opportunistic infections, presumably due to neutrophil reductions. If a serious infection develops, tocilizumab therapy should be stopped until the infection is controlled. In addition, treatments that suppress the immune system, such as tocilizumab, may cause an increase in the risk of cancer.
Drug Interactions9,10: Data from trials to date suggest that methotrexate, NSAIDs, or corticosteroids do not alter tocilizumab clearance. Tocilizumab has not been studied in combination with biologic DMARDs such as TNF antagonists.
Hepatic cytochrome enzymes are downregulated by infection and inflammation stimuli, including cytokines such as IL-6. Therefore, inhibition of IL-6 by tocilizumab may restore CYP450 activities to higher levels than those in the absence of tocilizumab, leading to increased metabolism of drugs that are substrates for these enzymes. In vitro studies showed that tocilizumab has the potential to affect expression of multiple CYP enzymes, including CYP1A2, CY2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. These effects may be clinically relevant for CYP450 substrates with narrow therapeutic indexes, where the dose is individually adjusted (e.g., warfarin, theophylline, cyclosporine). Thus, therapeutic monitoring for effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended upon initiation or discontinuation of tocilizumab. Caution should be exercised when tocilizumab is coadministered with CYP3A4 substrate drugs where decrease in effectiveness is undesirable, such as with oral contraceptives, lovastatin, and atorvastatin. The effect of tocilizumab on CYP450 enzyme activity may persist for several weeks after stopping therapy. Live vaccines should not be given concurrently with tocilizumab.
Dosage and Administration9: Tocilizumab is supplied as 20 mg/mL solutions for IV infusion in 80 mg/4 mL, 200 mg/10 mL, and 400 mg/20 mL single-use vials. When used in conjunction with a DMARD or as monotherapy, the recommended starting dose of tocilizumab is 4 mg/kg followed by an increase to 8 mg/kg based upon clinical response (not to exceed 800 mg per infusion). Repeat dosing of tocilizumab is recommended every 4 weeks. Dosage adjustments of tocilizumab are needed in patients with abnormal absolute neutrophil or platelet counts or elevated liver function tests. Tocilizumab should not be initiated in patients with an absolute neutrophil count <2,000 mm3 or a platelet count <100,000 mm3, or in those who have liver function tests greater than the ULN.
Ustekinumab (Stelara, Centocor Ortho Biotech)
Indication and Clinical Profile11,12: Ustekinumab, an interleukin antagonist given by SC injection, has been approved for treatment of adults with moderate-to-severe plaque psoriasis. It is the first agent in its class for this indication; the other biologic agents for psoriasis are T-cell or TNF inhibitors. Plaque psoriasis affects 6 million people in the U.S. and is an immune system disorder characterized by rapid overproduction of skin cells resulting in thickened patches of inflamed, red skin, often covered with silvery scales. Mild-to-moderate psoriasis is generally treated with topical corticosteroids (main therapy), calcitriol, calcipotriene, or tazarotene. Phototherapy is used when the disease is widespread or unresponsive to topical agents. Systemic drugs such as methotrexate, cyclosporine, and TNF inhibitors are usually reserved for moderate-to-severe disease.
The clinical development program, which was the basis for FDA approval of ustekinumab, included more than 2,200 patients in two pivotal phase III trials of 12 weeks’ duration. At the 12-week end point of both studies, at least two-thirds of patients who received two doses of ustekinumab, 45 mg or 90 mg, respectively, at weeks 0 and 4, showed 75% improvement on the Psoriasis Activity and Severity Index (PASI), compared with 3% to 4% in patients receiving placebo. A third 12-week trial in patients with moderate-to-severe psoriasis compared SC injections at weeks 0 and 4 of either 45 or 90 mg of ustekinumab with etanercept 50 mg twice weekly SC for 12 weeks. A PASI 75 response occurred in 67.5% of patients treated with 45 mg of ustekinumab, in 73.8% of those treated with 90 mg, and in 56.8% of those who received etanercept. Among the patients who did not respond to etanercept, 48.9% showed a PASI 75 response within 12 weeks after being switched to ustekinumab.
Pharmacology and Pharmacokinetics11: Ustekinumab is a human immunoglobulin G1 (IgG1) antibody directed against the p40 subunit of IL-12 and IL-23 cytokines, which are present in psoriasis skin lesions. These cytokines activate inflammatory and immune responses thought to be involved in the keratinocyte hyperplasia characteristic of psoriasis.
In clinical trials, the median tmax was 13.5 days and 7 days after a single SC administration of 45 mg and 90 mg doses, respectively. Following multiple SC doses, steady-state serum concentrations were achieved by week 28. There was no apparent accumulation in serum ustekinumab concentration over time when given SC every 12 weeks.
The metabolism of ustekinumab has not been characterized, but as a human IgG1-kappa monoclonal antibody, it is likely degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG. The mean half-life ranged from 15 to 46 days across all psoriasis studies following IV and SC administration. When given the same dose, subjects weighing >100 kg had lower median serum ustekinumab concentrations compared with those subjects weighing £100 kg. No pharmacokinetic data are available in patients with hepatic or renal impairment.
Adverse Reactions11,12: In clinical trials to date, adverse events occurred at similar rates in ustekinumab- and placebo-treated patients and were generally mild and self-limited. Based on its mechanism of action, however, ustekinumab may cause serious infections, including reactivation of latent tuberculosis. Thus, it should not be given to patients with any clinically important active infection, and patients should be evaluated for tuberculosis infection prior to initiating treatment. Moreover, blocking the p40 subunit of IL-12 may compromise tumor surveillance; doing so in mice led to a dramatic increase in the incidence of malignant tumors. Malignancies and reversible posterior leukoencephalopathy syndrome have been reported in patients treated with ustekinumab. Ustekinumab should be used during pregnancy or while nursing only if the potential benefit justifies the potential risk to the fetus (Pregnancy Category B).
Drug Interactions12: The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g., IL-1, IL-6, IL-10, TNF-alpha, interferon) during chronic inflammation. Thus, ustekinumab could normalize the formation of CYP450 enzymes. A role for IL-12 or IL-23 in the regulation of CYP450 enzymes has not been reported. However, upon initiation of ustekinumab in patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index, monitoring for therapeutic effect (e.g., warfarin) or drug concentration (e.g., cyclosporine) should be considered and the individual dose of the drug adjusted as needed. Live vaccines should not be given concurrently with ustekinumab. The safety of ustekinumab in combination with immunosuppressive agents or phototherapy has not been evaluated.
Dosage and Administration12: Ustekinumab is supplied in 45 mg/0.5 mL and 90 mg/1.0 mL single-use prefilled syringes and single-use vials for SC injection. For patients weighing £100 kg, the recommended dose is 45 mg initially and 4 weeks later, followed by 45 mg every 12 weeks. For patients weighing >100 kg, the recommended dose is 90 mg initially and 4 weeks later, followed by 90 mg every 12 weeks.
REFERENCES
1. Saphris (asenapine) package insert. Kenilworth, NJ: Schering-Plough (Merck & Co., Inc.); July 2009.
2. McIntyre RS, Cohen M, Zhao J, et al. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11:673-686.
3. Bepreve (bepotastine besilate) package insert. Irvine, CA: ISTA Pharmaceuticals, Inc; August 2009.
4. Protzko EE, Gomes PJ, Williams JI, et al. The ocular comfort and safety of the novel anti-histamine bepotastine besilate ophthalmic solution 1.5% in a healthy pediatric population [abstract]. J Allergy Clin Immunol. 2009;123:S50.
5. Multaq (dronedarone) package insert. Bridgewater, NJ: Sanofi-Aventis U.S. LLC; July 2009.
6. Davy JM, Herold M, Hoglund C, et al. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: the efficacy and safety of dronedarone for the control of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J. 2008;156:527.e1-e9.
7. Victoza (liraglutide injection) package insert. Princeton, NJ: Novo Nordisk Inc; January 2010.
8. Russell-Jones D, Vaag A, Schmitz O, et al. Liraglutide vs. insulin glargine and placebo in combination with metformin and sulfonylurea therapy in type 2 diabetes mellitus (LEAD-5 met+SU): a randomised controlled trial. Diabetologia. 2009;52:2046-2055.
9. Actemra (tocilizumab) package insert. South San Francisco, CA: Genentech, Inc; January 2010.
10. Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the TOWARD study. Arthritis Rheum. 2008;58:2968-2980.
11. Stelara (ustekinumab) package insert. Horsham, PA: Centocor Ortho Biotech Inc; December 2009.
12. Leonardi CL, Kimball A, Papp K, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results (PHOENIX 1). Lancet. 2008;371:1665-1674.
To comment on this article, contact rdavidson@uspharmacist.com.



