US Pharm. 2012;37(10):46-55.
ABSTRACT: New molecular entities (NMEs), as defined by the FDA, are drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2011–2012 ( TABLE 1) detail the basic clinical and pharmacologic profiles for each new drug, as well as its pharmacokinetics, adverse reactions, drug interactions, and dosing data. Note that the information for each NME was obtained primarily from sources published prior to FDA approval; thus, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature, such as the emergence of additional adverse reactions and black box warnings.
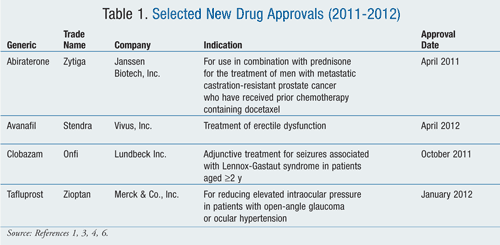
Abiraterone (Zytiga, Janssen Biotech, Inc.)
Indication and Clinical Profile1,2: Abiraterone acetate is the first oral, once-daily medication indicated for use in combination with prednisone for the treatment of men with metastatic castration-resistant prostate cancer (CRPC) who have received prior chemotherapy containing docetaxel.
Other than skin cancers, prostate cancer is the most frequently diagnosed cancer in men in the United States. In 2010, more than 217,000 new cases of prostate cancer were estimated and more than 32,000 men died from the disease. In prostate cancer, the male sex hormone testosterone stimulates prostate tumors to grow. Drugs or surgery are used to reduce testosterone production or to block testosterone’s effects. However, prostate cancer can sometimes continue to grow and metastasize beyond the prostate despite serum testosterone below castration levels. Men with these cancers are said to have CRPC.
FDA approval was based on a controlled, multicenter study involving 1,195 patients. These patients received either abiraterone orally at a dose of 1,000 mg once daily in combination with prednisone 5 mg orally twice daily or placebo once daily plus prednisone 5 mg orally twice daily. Treatment was administered until disease progression (defined as a 25% increase in prostate-specific antigen [PSA] over the patient’s baseline), initiation of new treatment, unacceptable toxicity, or withdrawal.
The prespecified interim analysis was conducted after 552 deaths and showed a statistically significant improvement in overall survival in subjects treated with abiraterone compared to placebo. An updated survival analysis was conducted when 775 deaths occurred. Results from this analysis were consistent with those from the interim analysis. In the interim analysis, the median survival was 14.8 months for the abiraterone arm and 10.9 months for the placebo arm. In the updated analysis, the median survival was 15.8 months and 11.2 months, respectively.
Pharmacology and Pharmacokinetics1,2: Abiraterone acetate is a pyridine-steroidal prodrug that is hydrolyzed in vivo to abiraterone (FIGURE 1). Abiraterone inhibits 17α-hydroxylase/C17,20-lyase, a CYP450 complex (CYP17) that is involved in testosterone production. This enzyme is expressed in testicular, adrenal, and prostatic tumor tissues and is required for androgen biosynthesis.
After oral administration, abiraterone reaches maximum concentrations (Cmax) in about 2 hours. Taking the drug with food results in a significant increase in Cmax and area under the curve (AUC). Abiraterone is partly metabolized in the liver by CYP3A4 to inactive metabolites and is excreted primarily in the feces, with a terminal half-life of about 12 hours.
Adverse Reactions and Drug Interactions1,2: The most commonly reported side effects (>5%) in patients receiving abiraterone include joint swelling or discomfort, hypo-kalemia, edema, muscle discomfort, hot flush, diarrhea, urinary tract infection (UTI), cough, hypertension, arrhythmia, urinary frequency, nocturia, dyspepsia, and upper respiratory tract infection. Abiraterone should be used with caution in patients with a history of cardiovascular disease or with medical conditions that might be compromised by increases in hypertension, hypokalemia, and fluid retention. Hypertension and hypokalemia should be corrected before and during treatment, and blood pressure, serum potassium, and symptoms of fluid retention should be monitored at least monthly.
CYP17 inhibition also causes adrenocortical insufficiency, hypertension, hypokalemia, and fluid retention. Coadministration of prednisone reduces the severity of these effects, which have led to drug interruption, dose modification, and/or discontinuation. Liver function should also be monitored since increases in liver enzymes have been reported.
Abiraterone is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6; thus, coadministration with CYP2D6 substrates that have a narrow therapeutic index should be avoided. If an alternative cannot be used, exercise caution and consider a dose reduction of the CYP2D6 substrate. Additionally, abiraterone is a substrate of CYP3A4 in vitro. Strong inhibitors and inducers of CYP3A4 should be avoided or used with caution.
Dosage and Administration1,2: Abiraterone is supplied as 250-mg tablets for oral administration. It should be taken with water and swallowed whole. Patients should not eat for 2 hours before taking the drug or for 1 hour afterwards. The recommended dosage is 1,000 mg administered orally once daily in combination with prednisone 5 mg administered orally twice daily.
The dosage should be decreased to 250 mg once daily in patients with moderate hepatic impairment; it should not be used in patients with severe hepatic impairment. If alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) increases to five times the upper limit of normal (ULN) or bilirubin increases to three times the ULN, the drug should be stopped and restarted at a lower dose.
Avanafil (Stendra, Vivus, Inc.)
Indication and Clinical Profile3: Avanafil is a new phosphodiesterase type 5 (PDE5) inhibitor approved to treat erectile dysfunction (ED). This common condition affects an estimated 30 million men in the U.S., and its incidence increases with age.
The drug’s safety and efficacy were established in three placebo-controlled clinical studies. A total of 1,923 patients were randomly assigned to the drug for up to 12 weeks at doses of 50 mg, 100 mg, or 200 mg, or to a placebo as needed about 30 minutes before sexual activity. The primary outcome measures were the erectile function domain of the International Index of Erectile Function (IIEF) and questions 2 and 3 from the Sexual Encounter Profile (SEP). Avanafil demonstrated statistically significant improvement from baseline in all three dose arms against placebo.
A subset of patients from two of these trials were enrolled in an extension trial of 3 months’ duration to evaluate avanafil (100 or 200 mg) in ED patients with type 1 or type 2 diabetes mellitus. In this study, patients in the avanafil treatment groups demonstrated statistically significant improvement from baseline against placebo based on the IIEF and SEP measures.
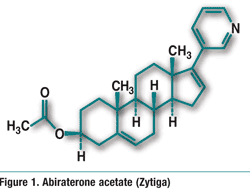
Pharmacology and Pharmacokinetics3: Avanafil is a highly selective pyrimidinecarboxamide PDE5 inhibitor (FIGURE 2). PDE5 is responsible for the degradation of cyclic guanosine monophosphate (cGMP), which produces smooth-muscle relaxation in the corpus cavernosum and allows inflow of blood. Erection of the penis involves release of nitric oxide (NO) in the corpus cavernosum during sexual stimulation. NO then activates the enzyme guanylate cyclase, which results in increased levels of cGMP, producing smooth-muscle relaxation in the corpus cavernosum and allowing inflow of blood. Avanafil enhances the effect of NO by inhibiting PDE5. Because sexual stimulation is required to initiate the local release of nitric oxide, the inhibition of PDE5 has no effect in the absence of sexual stimulation.
Oral avanafil is rapidly absorbed, and food does not significantly reduce bioavailability. The drug is extensively metabolized, primarily by CYP3A4, and the metabolites have significantly reduced ED activity. Avanafil is excreted as metabolites predominantly in the feces (62%) and to a lesser extent in the urine (21%). The drug has a terminal elimination half-life of approximately 5 hours.
Adverse Reactions and Drug Interactions3: The most common side effects reported in >2% of patients in the clinical studies of avanafil include headache, flushing, nasal congestion, nasopharyngitis, and back pain.
In rare cases, patients taking avanafil and other PDE5 inhibitors may get an erection lasting 4 hours or longer (priapism). When this occurs, patients should seek immediate medical care. In addition, PDE5 inhibitors rarely may cause color vision changes, even sudden loss of vision in one or both eyes and sudden loss or decrease in hearing. Patients who experience a sudden loss of vision or hearing should stop taking PDE5 inhibitors and immediately seek medical attention.
As with other PDE5 inhibitors, administration of avanafil is contraindicated with organic nitrates. Avanafil can potentiate the hypotensive effect of nitrates, alpha-blockers, antihypertensives, and alcohol. Avanafil is a CYP3A4 substrate that should not be used with strong CYP3A4 inhibitors and only used in reduced doses with moderate CYP3A4 inhibitors.
Dosage and Administration3: Avanafil is supplied as 50-, 100-, and 200-mg tablets for oral administration. The recommended initial dosage is 100 mg taken approximately 30 minutes before sexual activity with or without food. The dosage may be increased to 200 mg or decreased to 50 mg. The lowest dose that provides benefit should be used as needed no more than once per day. If used with a moderate CYP3A4 inhibitor, the dose should not exceed 50 mg in a 24-hour period. For patients on stable alpha-blocker therapy, the recommended starting dose is 50 mg.
Clobazam (Onfi, Lundbeck Inc.)
Indication and Clinical Profile4,5: Clobazam has been approved as an adjunctive treatment for seizures associated with Lennox-Gastaut syndrome (LGS) in patients 2 years of age and older. LGS is a severe form of epilepsy that usually begins before 4 years of age. It can be caused by a number of conditions, including brain malformations, severe head injuries, central nervous system (CNS) infections, and inherited degenerative or metabolic conditions. In 30% to 35% of patients, no cause can be found.
Patients commonly have frequent seizures of a wide variety, including tonic, atonic, atypical absence, and myoclonic. Most children with LGS experience some degree of impaired intellectual functioning or information processing, as well as developmental delays and behavioral disturbances. Since this disease affects fewer than 200,000 people in the U.S., clobazam was granted orphan drug designation by the FDA.
The efficacy of clobazam, when added to ongoing seizure medication, was established in two multicenter controlled studies of patients aged ≥2 years. In each study, the drug was tested for the amount of reduction in the weekly frequency of drop seizures (atonic, tonic, or myoclonic seizures resulting in a fall or loss of posture) from the 4-week baseline period to a maintenance period. In both studies, patients taking clobazam had improved seizure control when compared to those taking placebo or low-dose clobazam.
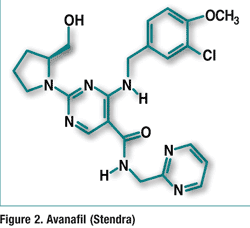
Pharmacology and Pharmacokinetics4,5: Clobazam is an antiepileptic drug of the 1,5-benzodiazepine class (FIGURE 3). The exact mechanism of action is not completely understood, although it likely involves potentiation of GABA-ergic neurotransmission, which results from binding at the benzodiazepine site of the GABAA receptor.
Clobazam is rapidly and extensively absorbed following oral administration and readily distributes to the CNS due to its lipophilicity. It is extensively metabolized in the liver, and the major metabolic pathway involves N-demethylation, primarily by CYP3A4 and to a lesser extent by CYP2C19 and CYP2B6.
N-desmethylclobazam, an active metabolite, is the major circulating metabolite in humans, and at therapeutic doses, plasma concentrations are three to five times higher than those of the parent compound. N-desmethylclobazam is extensively metabolized, primarily by CYP2C19. N-desmethyl-clobazam and its metabolites constitute approximately 94% of the total drug-related components in urine.
Adverse Reactions and Drug Interactions4,5: Common adverse reactions reported by patients taking clobazam include somnolence, sedation, fever, drooling, cough, constipation, insomnia, aggression, fatigue, UTI, upper respiratory tract infection, irritability, vomiting, dysphagia, ataxia, bronchitis, and pneumonia. Like other drugs of its class, clobazam should not be discontinued suddenly. Clobazam may slow thinking and impair motor skills; thus, patients should be cautioned against engaging in hazardous activities until its effects are known. It can also cause abuse and dependence and has been categorized as a Schedule IV drug.
As with other antiepileptics, clobazam may increase the risk of suicidal thoughts or behaviors in a small number of people taking the drug, so patients should be monitored for depression, suicidal thoughts or behavior, and unusual changes in mood or behavior. Clobazam is classified as a Pregnancy Category C drug.
CYP3A4 is the predominant enzyme that forms the active metabolite of clobazam, which is further metabolized by CYP2C19. Therefore, concurrent use of drugs that inhibit CYP2C19 (e.g., fluconazole or omeprazole) can increase serum concentrations of the active metabolite and possibly its toxicity. Similarly, patients with the “poor metabolizing” CYP2C19 polymorph may be at a greater risk for toxicity.
Clobazam inhibits CYP2D6; thus, drugs that are metabolized by 2D6 (e.g., fluoxetine or paroxetine) may require a dosage reduction if taken concomitantly with clobazam. Clobazam is also a mild inducer of CYP3A4 and may reduce serum concentrations of drugs metabolized by this pathway, including hormonal contraceptives.
Alcohol and clobazam have additive effects on CNS depression. In addition, alcohol can significantly increase plasma levels of clobazam.
Dosage and Administration4,5: Clobazam is supplied as 5-, 10-, and 20-mg tablets. The tablets can be administered whole or crushed and mixed in applesauce, and should be administered in divided doses twice daily. The recommended starting dosage for patients who weigh ≤30 kg is 5 mg once daily for at least 1 week. The dose can be titrated to 5 mg twice daily and then to 10 mg twice daily; the patient should take each dose for at least 1 week before increasing it. For patients weighing >30 kg, the recommended initial dosage is 5 mg twice daily for at least 1 week, followed by an increase to 10 mg twice daily and then to a maintenance dose of 20 mg twice daily.
Patients ≥65 years, those with mild-to-moderate hepatic impairment, and those who are poor CYP2C19 metabolizers should take 5 mg once daily initially. To avoid withdrawal symptoms or exacerbation of seizures when discontinuing the drug, the daily dose should be reduced weekly by 5 to 10 mg.
Tafluprost (Zioptan, Merck & Co., Inc.)
Indication and Clinical Profile6,7: Glaucoma is an eye disease associated with increased fluid pressure in the eye, eventually causing permanent damage to the optic nerve, impairing the field of vision, and ultimately leading to blindness. Glaucomas are subclassified as open-angle glaucoma (OAG) and closed-angle glaucoma (CAG). CAG is an acute disease with rapid progression, while OAG is a slowly progressive disease responsible for about 90% of glaucoma cases in the U.S. Management of OAG typically includes medication to lower intraocular pressure (IOP) and surgery.
Tafluprost is the newest ophthalmic prostaglandin approved for reducing elevated IOP in patients with OAG or ocular hypertension. It is an analogue of prostaglandin F2α, similar to other antiglaucoma drugs. Prostaglandin analogues, including latanoprost, bimatoprost, and travo-prost, are often used as first-line treatment to lower IOP in patients with OAG. However, unlike other prostaglandin drugs, tafluprost is preservative free; it does not contain benzalkonium chloride, which may be irritating to some patients’ eyes.
The FDA approval of tafluprost was based on results from five controlled clinical studies of up to 2 years in 905 patients. Both preservative-containing and preservative-free formulations of tafluprost were used. In clinical studies of up to 2 years in duration, tafluprost, dosed once daily in the evening, lowered IOP at 3 and 6 months by 6 to 8 mmHg and 5 to 8 mmHg, respectively, from a baseline pressure of 23 to 26 mmHg. Tafluprost, under other trademarks, has been marketed outside the U.S. since 2008.
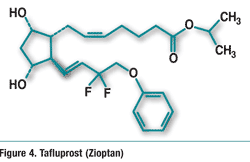
Pharmacology and Pharmacokinetics6,7: Tafluprost is a fluorinated analogue of prostaglandin F2α and an ester prodrug (FIGURE 4). Ester-ification facilitates corneal permeation. Esterases in the eye convert the drug to the active prosta-glandin acid form. The active acid is a selective FP prostanoid receptor agonist that is believed to reduce IOP by increasing uveoscleral outflow. The exact mechanism of action is unknown at this time. Mean plasma Cmax is about 26 to 27 pg/mL, and mean plasma AUC is 394 to 432 pg*min/mL.
Adverse Reactions and Drug Interactions6,7: In clinical trials of patients receiving either preservative-containing or preservative-free tafluprost, the most common (4%-20%) pooled adverse reaction observed was conjunctival hyperemia. Tafluprost may gradually change eyelashes and vellus hair in the treated eye. These changes include increased length, color, thickness, shape, and number of lashes and are usually reversible upon discontinuation of treatment.
Tafluprost also has been reported to cause increased pigmentation of the iris, periorbital tissue (eyelid), and eyelashes. After discontinuation, pigmentation of the iris is likely to be permanent, while pigmentation of the periorbital tissue and eyelash changes have been reported to be reversible in some patients.
Tafluprost should be used with caution in patients with active intraocular inflammation because the inflammation may be exacerbated. Macular edema, including cystoid macular edema, has been reported during treatment with prostaglandin F2α analogues. Tafluprost should also be used with caution in aphakic patients (i.e., lacking a lens in the eye), in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Tafluprost should not be used during pregnancy unless the potential benefit justifies the potential risk to the fetus, and caution should be exercised when it is administered during nursing (Pregnancy Category B). No significant drug interactions are anticipated.
Dosage and Administration6,7: Tafluprost is provided in 0.3 mL single-use containers containing 0.015 mg/mL of the drug. The product does not contain preservatives, is a sterile solution for single use only, and is of sufficient quantity to treat one or both eyes. The recommended dosage is one drop in the conjunctival sac of the affected eye(s) once daily in the evening. The dose should not exceed once daily, as more frequent administration of prostaglandin analogues may lessen the IOP-lowering effect. Since tafluprost does not contain preservatives, any unused solution should be discarded immediately.
Tafluprost may be used concomitantly with other ophthalmic products that lower IOP. If more than one topical ophthalmic product is being used, they should be administered at least 5 minutes apart from each other.
REFERENCES
1. Zytiga (abiraterone acetate) package insert. Horsham, PA: Janssen Biotech, Inc; April 2011.
2. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
3. Stendra (avanafil) package insert. Mountain View, CA: Vivus, Inc; April 2012.
4. Onfi (clobazam) package insert. Deerfield, IL: Lundbeck Inc; October 2011.
5. Ng YT, Conry JA, Drummond R, et al; OV-1012 Study
Investigators. Randomized, phase III study results of clobazam in
Lennox-Gastaut syndrome. Neurology. 2011;77:1473-1481.
6. Zioptan (tafluprost) package insert. Whitehouse Station, NJ: Merck & Co., Inc; January 2012.
7. Erb C, Lanzl I, Seidova SF, Kimmich F.
Preservative-free tafluprost 0.0015% in the treatment of patients with
glaucoma and ocular hypertension. Adv Therapy. 2011;28:575-585.
To comment on this article, contact rdavidson@uspharmacist.com.



