US Pharm. 2013;38(10):27-34.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2012–2013 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction (AR), drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s new drug application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” ARs for many NMEs within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one Black Box Warning for serious ARs or are withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.

Alogliptin (Nesina, Takeda Pharmaceuticals America)
Indication and Clinical Profile1,2: Alogliptin is a new dipeptidyl peptidase-4 (DPP-4) inhibitor approved as an adjunct to diet and exercise to improve glycemic control in patients with type 2 diabetes mellitus (DM2). DM2 is a chronic, progressive disease affecting more than 23 million people in the United States. Even with diet and exercise, patients often must take multiple medications to help manage their blood glucose levels. Alogliptin is the fifth DPP-4 inhibitor approved by the FDA since 2006 (the others being linagliptin, saxagliptin, vildagliptin, and sitagliptin).
Approval of alogliptin was based on three 26-week, double-blind, randomized, placebo- and active-controlled studies involving more than 1,768 patients with inadequate glycemic control through diet and exercise. Alogliptin 25 mg once daily was superior to placebo in improvements from baseline to week 26 for glycosylated hemoglobin (A1C) and fasting plasma glucose (FPG). Compared with the respective monotherapies, alogliptin coadministered with either pioglitazone or metformin produced significant improvements in A1C and FPG from baseline. Based on these studies, alogliptin also was approved in combination with pioglitazone (as Oseni) and metformin HCl (as Kazano).
Pharmacology and Pharmacokinetics1,2: Alogliptin is a dihydropyrimidinedione (FIGURE 1) that functions as a DPP-4 inhibitor. DPP-4 is involved in the inactivation of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), each of which enables glucose-dependent insulin secretion from the pancreas. By functioning as a DPP-4 inhibitor, alogliptin slows GLP-1 and GIP inactivation, thereby reducing blood glucose levels.
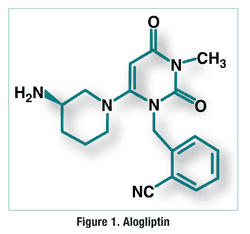
The absolute bioavailability of alogliptin is 100% when the drug is taken with or without food. Alogliptin has a peak plasma concentration of 1 to 2 hours and a half-life of 21 hours. It undergoes minimal CYP450 metabolism, resulting in formation of an active N-demethylated metabolite (1%) and some conjugation yielding an inactive metabolite. Alogliptin does not appear to significantly induce or inhibit metabolism by any major cytochrome isozyme. Alogliptin is eliminated primarily via renal excretion (76%), along with some fecal elimination (13%).
Adverse Reactions (ARs) and Drug Interactions1,2: The most common ARs reported in clinical trials were nasopharyngitis, headache, and upper respiratory tract infection. Alogliptin produced hypoglycemia at a rate comparable to that for placebo. There have been postmarketing reports of acute pancreatitis, severe hypersensitivity reactions (anaphylaxis, angioedema, and cutaneous ARs), and hepatic failure (sometimes fatal) in patients taking alogliptin. Alogliptin is not predicted to increase the risk of fetal developmental abnormalities, but in the absence of more concrete data, consideration should be given to whether the potential benefits of treatment outweigh the potential risks (Pregnancy Category B).
Alogliptin has no significant interactions with drugs that are CYP substrates or inhibitors, or with drugs excreted renally. Patients taking insulin or an insulin secretagogue in combination with alogliptin may have to lower the insulin or insulin secretagogue dosage to reduce the risk of hypoglycemia.
Dosage and Administration1,2: Alogliptin is supplied as 6.25-mg, 12.5-mg, and 25-mg tablets. The recommended dosage is 25 mg once daily, with or without food. Patients with moderate renal impairment should take 12.5 mg once daily, and those with severe renal impairment or end-stage renal disease should take 6.25 mg once daily. No initial dosage adjustment is recommended for patients who are elderly or who have mild renal or mild-to-moderate hepatic impairment. Alogliptin has not been studied in patients with severe hepatic impairment. Alogliptin is also available as combination therapy under the names Kazano (alogliptin + metformin HCl) and Oseni (alogliptin + pioglitazone).
Tofacitinib (Xeljanz, Pfizer)
Indication and Clinical Profile3,4: Tofacitinib, a novel Janus kinase (Jak) 3 inhibitor, is approved to treat adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or are intolerant to methotrexate (MTX). Ruxolitinib (Jakafi), the only other Jak inhibitor available in the United States, is approved to treat myelofibrosis. RA, an autoimmune disease that causes inflammation of the joints and surrounding tissues, affects about 1.5 million Americans. Initial treatment typically includes a disease-modifying antirheumatic drug (DMARD) and a nonsteroidal anti-inflammatory drug or corticosteroid to control symptoms. MTX is generally the DMARD of choice; hydroxychloroquine is a safer alternative that may be appropriate in milder cases. Tumor necrosis factor inhibitors (TNFIs) are usually the first-line biologic agents prescribed after inadequate response to a DMARD; they may be given as monotherapy or in combination with MTX. For patients with an inadequate response to a TNFI, it may be effective to switch to a second TNFI or to a non-TNF biologic agent with a different mechanism of action. Tofacitinib is a new biologic treatment option for adults with RA who have a poor response to MTX. It may be used as monotherapy or in combination with MTX or other nonbiologic DMARDs.
FDA approval of tofacitinib was based on two dose-ranging trials and five confirmatory trials in more than 3,300 patients with moderate-to-severe RA who did not respond to previous treatment with MTX or another nonbiologic DMARD, or with a TNFI or another biologic DMARD. In all trials, compared with patients taking placebo, patients taking tofacitinib 5 mg or 10 mg twice daily—with or without background DMARD treatment—had higher ACR20, ACR50, and ACR70 response rates (RRs) (20%, 50%, and 70% improvement on the American College of Rheumatology [ACR] scale, respectively) at months 3 and 6. Higher ACR20 RRs were observed within 2 weeks versus placebo. In the 12-month trials, ACR RRs in tofacitinib-treated patients were consistent at 6 and 12 months. In these trials, a greater proportion of patients taking tofacitinib 5 mg or 10 mg twice daily plus MTX achieved a low level of disease activity, as measured by a Disease Activity Score (DAS28-4 [erythrocyte sedimentation rate]) <2.6, at 6 months, versus patients taking MTX alone; they also showed significant improvement on the Health Assessment Questionnaire Disability Index.
Pharmacology and Pharmacokinetics3,4: Tofacitinib is a pyrrolopyrimidine (FIGURE 2) that acts as a Jak3 inhibitor. Jaks are intracellular enzymes that transmit signals arising from cytokine or growth factor receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune-cell function. Within the signaling pathway, Jaks phosphorylate and activate signal transducers and activators of transcription (STATs), which modulate intracellular activity, including gene expression. Tofacitinib modulates the signaling pathway at the point of Jaks, preventing phosphorylation and activation of STATs. In a murine model of established arthritis, tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. The efficacy of this disease model correlated with inhibition of both Jak1 and Jak3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to Jak3 inhibition.
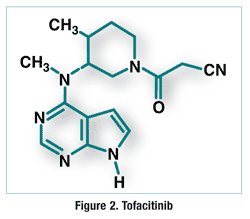
Adverse Reactions (ARs) and Drug Interactions3,4: The most common ARs associated with tofacitinib in clinical trials were upper respiratory tract infections, headache, diarrhea, nasopharyngitis, and hypertension. Tofacitinib also was associated with an increased risk of serious infections, including tuberculosis (TB) and other opportunistic infections, and with lymphoma and other cancers. The product labeling has a Black Box Warning regarding these safety risks. Patients should be tested for latent or active TB before starting tofacitinib. Eleven solid cancers and one lymphoma were diagnosed in 3,328 patients taking tofacitinib with or without a DMARD for ≤12 months, versus no solid cancers or lymphoma in 809 patients taking placebo with or without a DMARD for 3 to 6 months. The significance of this finding is unclear.
Tofacitinib treatment is associated with gastrointestinal perforation (especially in patients with diverticulitis), increases in cholesterol and liver-enzyme tests, and reductions in blood counts (lymphocytopenia, neutropenia, hemoglobin levels). Tofacitinib was approved with a Risk Evaluation and Mitigation Strategy, which consists of a Medication Guide advising patients about important safety information and a communication plan informing health care providers about the serious risks associated with tofacitinib and advising regular monitoring. The FDA is also requiring a postmarketing study to evaluate the long-term effects of tofacitinib with regard to heart disease, cancer, and serious infections. Tofacitinib has demonstrated feticidal and teratogenic effects in animals and is classified as Pregnancy Category C.
Tofacitinib is metabolized by CYP3A4. Thus, potent inhibitors of CYP3A4, (e.g., ketoconazole) can increase serum concentrations of tofacitinib, and the tofacitinib dosage should be reduced to 5 mg once daily if these drugs are taken concurrently. Drugs that are both moderate CYP3A4 inhibitors and potent CYP2C19 inhibitors (e.g., fluconazole) also increase serum concentrations of tofacitinib; concurrent use requires a reduction in tofacitinib dosage. Potent inducers of CYP3A4 (e.g., rifampin) decrease tofacitinib levels and should be avoided. Tofacitinib should not be coadministered with other biologic agents or potent immunosuppressive drugs. Live vaccines should not be given to patients taking tofacitinib; immunizations should be updated before tofacitinib is initiated.
Dosage and Administration3,4: Tofacitinib is supplied as 5-mg tablets. The recommended initial dosage is 5 mg twice daily, with or without food. The dosage should be reduced to 5 mg once daily in patients with moderate hepatic impairment or moderate-to-severe renal impairment. The drug is not recommended for patients with severe hepatic impairment or for those with a low lymphocyte count (<500 cells/mm3), a low absolute neutrophil count (<1,000 cells/mm3), or low hemoglobin levels (<9 g/dL).
Apixaban (Eliquis, Bristol-Myers Squibb, Pfizer)
Indication and Clinical Profile5,6: Apixaban is the newest anticoagulant approved by the FDA to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (AF). AF, which affects approximately 3 million people in the United States, causes the heart to beat with less strength and regularity, resulting in blood pooling in the atria. This results in a greater likelihood of clot formation, which increases the risk of stroke and systemic embolism. Anticoagulant therapy is recommended for patients with AF and a CHADS2 (congestive heart failure, hypertension, age ≥75 years, diabetes mellitus, or stroke × 2) score ≥1. For years, the anticoagulant of choice for AF patients has been warfarin; however, warfarin has multiple drug and food interactions and requires regular international normalized ratio (INR) monitoring. Apixaban is the third anticoagulant since 2010 to be approved as an alternative to warfarin (the others are rivaroxaban and dabigatran) for patients who are nonadherent to routine INR monitoring or did not achieve clinical benefit from warfarin.
Approval of apixaban was based on two 2-year, multicenter, randomized, head-to-head, double-blind studies that involved more than 23,000 patients with AF. These studies demonstrated that, at endpoint, apixaban was statistically superior to placebo and active comparators (warfarin and aspirin) for reducing stroke and systemic embolism in patients who have nonvalvular AF.
Pharmacology and Pharmacokinetics5,6: Apixaban is a pyrazolopyridinone (FIGURE 3) that indirectly decreases platelet aggregation by reversibly inhibiting free and clot-bound factor Xa (FXa) at the active site. FXa antagonism results in reduced prothrombinase activity, causing a decrease in thrombin generation. This mechanism inhibits development of thrombi that are induced by thrombin.
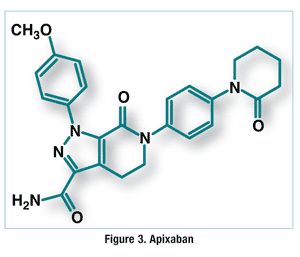
The estimated absolute bioavailability of apixaban is 50% when the drug is taken with or without food. Apixaban undergoes prolonged absorption, with a half-life of 12 hours, and it is approximately 87% plasma protein bound. Apixaban is metabolized by O-demethylation and hydroxylation to inactive metabolites, mainly by CYP3A4, with minor contributions from other cytochrome isozymes. The drug is eliminated in the urine and feces, 25% of it as inactive metabolites.
Adverse Reactions (ARs) and Drug Interactions5,6: In clinical trials, ARs were similar to those reported in patients taking other anticoagulants. The most frequent ARs reported in clinical trials were increased bleeding and symptoms of increased bleeding (paleness, fatigue). Major bleeding events (intracranial, intraspinal, intraocular, pericardial, intra-articular, intramuscular, severe retroperitoneal), some of them fatal, have occurred; the risk is increased in patients aged >75 years and in patients whose INR with warfarin was within goal (2.0-3.0). Any patient exhibiting signs of active pathologic bleeding should not take apixaban. Apixaban increases the risk of bleeding during labor and pregnancy; therefore, in pregnant patients, consideration should be given to whether the potential benefits outweigh the potential risks (Pregnancy Category B). Apixaban should not be taken by patients who are breastfeeding, as it is currently unknown whether the drug is excreted in breast milk. The safety and efficacy of apixaban have not been established in patients with prosthetic heart valves, so the drug should be avoided in this population.
Coadministration of drugs affecting hemostasis (selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, fibrinolytics, heparin, aspirin, nonsteroidal anti-inflammatory drugs) will result in an increased risk of bleeding. The apixaban dosage should be reduced when the drug is coadministered with strong dual inhibitors of CYP3A4 and P-glycoprotein (Pgp) (ketoconazole, itraconazole, ritonavir, clarithromycin). Concomitant use of apixaban with strong dual inducers of CYP3A4 and Pgp (rifampin, carbamazepine, phenytoin, St. John’s wort) should be avoided, as this can result in inadequate drug concentrations. Procoagulant reversal agents (prothrombin complex concentrate, recombinant factor VIIa) may potentially reverse the effects of apixaban, but this has not been evaluated.
Dosage and Administration5,6: Apixaban is available as capsules of 2.5 mg and 5 mg. The recommended dosage for most patients is 5 mg twice daily. No dosage adjustment is required for patients with mild hepatic impairment, but because of insufficient clinical data, a dosage adjustment for moderate hepatic impairment is not available. Apixaban should be reduced to 2.5 mg twice daily when coadministered with strong dual inhibitors of CYP3A4 and Pgp or when used in patients with at least two of the following characteristics: age >80 years, body weight <60 kg, and serum creatinine >1.5 mg/dL.
Apixaban should be temporarily discontinued ≥24 hours prior to surgery or invasive procedures with a low risk of bleeding, and 48 hours prior to procedures with a moderate or high risk of unacceptable or clinically significant bleeding. Apixaban should not be permanently discontinued without starting an alternative anticoagulant, as doing so incurs an increased risk of stroke or other thrombotic events.
Perampanel (Fycompa, Eisai)
Indication and Clinical Profile7,8: Perampanel is approved to treat partial-onset seizures in epilepsy patients aged ≥12 years. Partial-onset seizures, the most common type of seizure in patients with epilepsy, affect only a limited or localized area of the brain at onset, but they can spread to other parts of the brain. Seizures cause a wide range of symptoms, including repetitive limb movements (spasms), unusual behavior, and generalized convulsions with loss of consciousness. Many epilepsy patients do not achieve satisfactory seizure control with the treatments currently available; thus, it is important to have a variety of options.
FDA approval of perampanel was based on three randomized, double-blind, placebo-controlled, multicenter trials involving 1,037 patients aged ≥12 years. All trials had a 6-week baseline period during which patients needed to have more than five seizures in order to be randomized. Median baseline seizure frequency ranged from 9.3 to 14.3 seizures per 28 days. The baseline period was followed by a 19-week treatment period consisting of a 6-week titration phase and a 13-week maintenance phase. The primary endpoint in all trials was the percentage change in seizure frequency per 28 days during the treatment period compared with the baseline period. A statistically significant decrease in seizure rate was observed at dosages of 4 mg to 8 mg/day, with little additional reduction in frequency at 12 mg/day. Proportions of patients with a ≥50% reduction in seizure frequency were 19.3%, 28.5%, 35.3%, and 35.0% for placebo, 4 mg, 8 mg, and 12 mg, respectively.
Pharmacology and Pharmacokinetics7,8: Perampanel is a pyridopyridone (FIGURE 4) that acts as a selective, noncompetitive antagonist of the ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptor on postsynaptic central nervous system (CNS) neurons. Glutamate is the primary excitatory neurotransmitter in the CNS and is implicated in a number of neurologic disorders caused by neuronal overexcitation. Perampanel has high potency and a prolonged terminal half-life of approximately 70 hours. The drug is 95% bound to plasma protein, and its primary route of metabolism is via CYP3A4. Perampanel does not induce or inhibit any CYP isoenzymes. About 70% of the dose is excreted in the feces, and 30% is excreted in the urine; less than 2% is excreted unchanged in the urine.
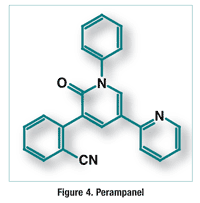
Adverse Reactions (ARs) and Drug Interaction7,8: In clinical trials, the most common ARs reported with perampanel were dizziness, drowsiness, fatigue, irritability, falls, upper respiratory tract infection, weight increase, vertigo, ataxia, gait disturbance, balance disorder, anxiety, blurred vision, dysarthria, asthenia, aggression, and hypersomnia. The drug has a Black Box Warning concerning the risk of serious neuropsychiatric events, including irritability, aggression, anger, anxiety, paranoia, euphoric mood, agitation, and mental status changes. Some of these events were serious and life-threatening. Health care providers should be alerted immediately if changes in mood or behavior that are atypical for the patient are observed. Patients should also be closely monitored during the titration period, when higher dosages are used.
Dosage and Administration7,8: Perampanel is supplied as 2-, 4-, 6-, 8-, 10-, and 12-mg tablets. The minimum effective dosage is 4 mg once daily. Dosages of 8 mg and 12 mg confer a greater therapeutic benefit, but also an increase in ARs. In the absence of enzyme-inducing antiepileptic drugs (AEDs), the recommended starting dose is 2 mg once daily at bedtime. The dosage is then increased by increments of 2 mg/day, no more frequently than every week, to a total of 4 mg to 8 mg once daily at bedtime. In elderly patients, dosage increases during titration are recommended no more frequently than every 2 weeks. The recommended maximum dosage is 12 mg once daily. In the presence of enzyme-inducing AEDs (e.g., phenytoin, carbamazepine, oxcarbazepine), the recommended starting dose is 4 mg, and patients should be monitored closely for response.
Pasireotide (Signifor, Novartis)
Indication and Clinical Profile9,10: Pasireotide diaspartate is specifically indicated for the treatment of Cushing’s disease in adults for whom pituitary surgery is not an option or has not been curative. In Cushing’s disease, a tumor in the pituitary gland leads to overproduction and release of adrenocorticotropic hormone (ACTH), resulting in hyperstimulation of the adrenal gland and overproduction of cortisol. Cortisol regulates many important bodily functions, including response to stress and injury. Patients with Cushing’s disease may have increased weight, glucose intolerance or diabetes, high blood pressure, easy bruising, and increased risk of infections. Although pituitary surgery tends to be first-line therapy for Cushing’s disease, it is not always successful, and some patients refuse to undergo the procedure.
The safety and effectiveness of pasireotide were evaluated in a multicenter, randomized clinical trial involving 162 patients. Most patients had persistent or recurrent Cushing’s disease despite pituitary surgery; the rest were de novo patients for whom surgery was not indicated or who had refused surgery. Patients were assigned to one of two pasireotide regimens (0.6 mg subcutaneously [SC] twice daily [bid] or 0.9 mg SC bid) for a 6-month period. The dosage could be reduced by 0.3 mg bid for intolerability at any time, and some patients who safely responded to the medication were allowed to continue treatment. The primary efficacy endpoint was the proportion of patients who achieved normalization of mean 24-hour urinary free cortisol (UFC) levels after 6 months and did not dose-increase during this period. At month 6, percentages of responders for the primary endpoint were 15% and 26% in the 0.6 mg bid and 0.9 mg bid groups, respectively, with some reductions observed as early as 1 month after treatment initiation. For patients who remained in the trial (n = 78), similar UFC-lowering was observed at the end of 1 year.
Pharmacology and Pharmacokinetics9,10: Pasireotide is a somatostatin-based cyclohexapeptide somatostatin analogue (FIGURE 5) that exerts its pharmacologic activity via binding to somatostatin receptor (SSTS) subtypes 1 through 5. In Cushing’s disease, pituitary corticotroph tumor cells frequently overexpress SSTS-5, whereas the other receptor subtypes often are not expressed or are expressed at lower levels. Pasireotide has a 40-fold increased affinity for SSTS-5, and binding to this receptor leads to inhibition of ACTH secretion, which in turn results in reduced adrenal cortisol secretion.
Pasireotide gives peak plasma concentrations within 30 minutes postadministration and is widely distributed. It is not significantly metabolized, but somatostatin analogues (including pasireotide) may decrease the metabolic clearance of compounds metabolized by CYP450 enzymes. Pasireotide is a substrate for P-glycoprotein pumps, but no other known transporter. The drug is cleared primarily hepatically and appears to have a half-life of about 12 hours.
Adverse Reactions (ARs) and Drug Interactions9,10: The most common ARs observed in clinical trials with pasireotide were hyperglycemia, diarrhea, nausea, abdominal pain, and gallstones. Hyperglycemia could be detected as early as 2 weeks after treatment initiation, and continued treatment caused or worsened diabetes in some patients. Therefore, patients should be carefully monitored and treated appropriately with antidiabetic therapies, including insulin. Also, based on its mechanism, pasireotide may cause biochemical and/or clinical hypocortisolism. If this occurs, it may be necessary to reduce or interrupt the dosage and/or add a low-dose short-term glucocorticoid.
The FDA is requiring several postmarketing studies for pasireotide that focus on safety monitoring for serious hyperglycemia, acute liver injury, and adrenal insufficiency. Pasireotide is Pregnancy Category C, and it should not be used while nursing since it is excreted in breast milk. The safety and effectiveness of pasireotide have not been established in pediatric patients.
Concurrent use of cyclosporine with pasireotide may decrease the relative bioavailability of cyclosporine; therefore, dosage adjustment of cyclosporine to maintain therapeutic levels may be necessary. Caution is required when pasireotide is coadministered with antiarrhythmic agents and other drugs that may prolong the QT interval. No significant interactions have been observed between pasireotide and drugs used to manage hyperglycemia (metformin, nateglinide, liraglutide).
Dosage and Administration9,10: Pasireotide is supplied as a solution for SC injection. The recommended initial dose is either 0.6 mg or 0.9 mg SC twice daily. Patients should be evaluated for treatment response, including a clinically significant reduction in 24-hour urinary free cortisol levels and/or improvement in disease signs or symptoms. Patients should continue pasireotide therapy as long as benefit is derived. The drug is dispensed with a Medication Guide, including instructions for patients and caregivers that describe the risks and ARs to be mindful of when using pasireotide. See the prescription label for any dosing modifications.
REFERENCES
1. Nesina (alogliptin) product information. Deerfield, IL: Takeda Pharmaceuticals America, Inc; January 2013.
2. DeFronzo RA, Fleck PR, Wilson CA, Mekki Q; Alogliptin Study 010
Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor
alogliptin in patients with type 2 diabetes and inadequate glycemic
control: a randomized, double-blind, placebo-controlled study. Diabetes Care. 2008;31:2315-2317.
3. Xeljanz (tofacitinib) product information. New York, NY: Pfizer Inc; November 2012.
4. Fleischmann R, Kremer J, Cush J, et al; ORAL Solo Investigators.
Placebo-controlled trial of tofacitinib monotherapy in rheumatoid
arthritis. N Engl J Med. 2012;367:495-507.
5. Eliquis (apixaban) product information. Princeton, NJ: Bristol-Myers Squibb Co, and New York, NY: Pfizer Inc; December 2012.
6. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees
and Investigators. Apixaban versus warfarin in patients with atrial
fibrillation. N Engl J Med. 2011;365:981-992.
7. Fycompa (perampanel) product information. Woodcliff Lake, NJ: Eisai Inc; October 2012.
8. French JA, Krauss GL, Biton V, et al. Adjunctive perampanel for
refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79:589-596.
9. Signifor (pasireotide diaspartate) product information. East Hanover, NJ: Novartis Pharmaceuticals Corp; December 2012.
10. Colao A, Petersenn S, Newell-Price J, et al; Pasireotide B2305
Study Group. A 12-month phase 3 study of pasireotide in Cushing’s
disease. N Engl J Med. 2012;366:914-924.
To comment on this article, contact rdavidson@uspharmacist.com.



