US Pharm. 2016;41(10):30-36.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2015–2016 (TABLE 1) detail the basic clinical and pharmacologic profiles for each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Insulin Degludec (Tresiba, Novo Nordisk)
Indication and Clinical Profile1,2: Insulin degludec is a long-acting insulin analogue indicated for improving glycemic control in adults with type 1 or type 2 diabetes (T1D, T2D). The CDC estimates that 29 million people in the United States have been diagnosed with diabetes. Over time, diabetes increases the risk of serious health complications, including heart disease, blindness, neuropathies, and nephropathy. Improvement in blood-sugar control can reduce the risk of some of these long-term complications. Long-acting insulins play an essential role in the treatment of patients with T1D and in T2D patients with advanced disease.
The efficacy and safety of insulin degludec for the treatment of patients with T1D and T2D were evaluated in several controlled trials. Insulin degludec was studied as an alternative to insulin glargine as part of a basal-bolus regimen in the BEGIN Basal-Bolus Type 1 trial, which included 629 patients with T1D. Patients were randomized in a 3:1 ratio to either insulin degludec (n = 472) or insulin glargine (n = 157) plus mealtime insulin aspart. Patients in the degludec treatment arm were switched from basal insulin to insulin degludec in a 1:1 ratio, with a 20% to 30% dose reduction in patients receiving multiple basal doses per day. After 52 weeks, the reduction in HbA1c patients treated with insulin degludec was similar to that in patients treated with insulin glargine (0.40% vs. 0.39%), meeting the criteria for noninferiority. However, rates of nocturnal hypoglycemia were 27% lower in patients receiving insulin degludec versus insulin glargine (3.91% vs. 5.22%; P = .024).
In the BEGIN Basal-Bolus Type 2 trial, insulin degludec was studied as an alternative to insulin glargine in 1,006 patients with T2D. Patients were randomized to receive either insulin degludec or insulin glargine plus mealtime insulin aspart, metformin, and/or pioglitazone. Patients had an average HbA1c of 8.3% to 8.4%, and 50% of them were on a regimen of basal-bolus insulin and oral antidiabetic medications. After 52 weeks, insulin degludec was found to be noninferior to insulin glargine, providing a similar HbA1c-lowering effect (–1.10% vs. –1.18%). However, overall rates of hypoglycemia were significantly lower with insulin degludec (11.09% vs. 13.63%; P = .0359), as were rates of nocturnal hypoglycemia (1.39% vs. 1.84%; P = .0399).
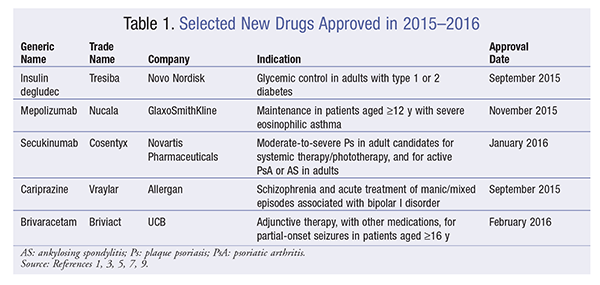
Pharmacology and Pharmacokinetics1,2: Insulin degludec is a modified human insulin in which a single amino acid is deleted and a conjugated hexadecanedioic acid is added via a gamma-L-glutamyl spacer at the amino acid lysine, at position B29 (FIGURE 1). The hexadecanedioic acid allows the formation of multihexamers in subcutaneous (SC) tissues following administration, creating an SC depot that results in slow insulin release to the systemic circulation. Also, in the circulation, insulin degludec is bound by plasma albumin, further extending its duration. Therefore, insulin degludec has a slow onset of action (30-90 minutes)—similar to that of insulin glargine and detemir—and no peak in activity because of the slow diffusion to the systemic circulation. The duration of action of insulin degludec is reported to be as high as 42 hours, compared with 18 to 26 hours provided by other long-acting insulins (glargine and detemir), making it a once-daily basal insulin. No clinically relevant pharmacokinetic differences were observed in patients with renal or hepatic impairment who received insulin degludec. All insulin products lower blood glucose by stimulating peripheral glucose uptake, especially by skeletal muscle and fat, and by inhibiting hepatic glucose production. Insulins also inhibit lipolysis and proteolysis and enhance protein synthesis.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions associated with insulin degludec in clinical trials were hypoglycemia, allergic reactions, injection-site reactions (including lipodystrophy), itching, rash, edema, and weight gain. Severe, life-threatening, generalized allergy (including anaphylaxis), generalized skin reactions, angioedema, bronchospasm, hypotension, and shock may occur with any insulin, as may hypokalemia. It may be necessary to adjust insulin degludec doses in patients in whom other drugs that affect glucose metabolism are added. Also, concurrent antiadrenergic drugs may mask the signs and symptoms of hypoglycemia. Insulin degludec should not be used in patients with diabetic ketoacidosis.
Dosage and Administration1,2: Insulin degludec is supplied as an injection for SC administration in FlexTouch pens containing 100 or 200 U/mL of insulin. It should be injected SC once daily, at any time of day, into the thigh, upper arm, or abdomen. To reduce the risk of lipodystrophy, injection sites within the same region should be rotated from one injection to the next. Doses should be individualized based on the patient’s metabolic needs, blood glucose–monitoring results, and glycemic-control goal. Patients with changes in insulin dosage, coadministration of other glucose-lowering medications, meal patterns, and physical activity, as well as patients with renal or hepatic impairment or hypoglycemia unawareness, should be monitored more closely.
Mepolizumab (Nucala, GlaxoSmithKline)
Indication and Clinical Profile3,4: Mepolizumab has been approved for use as an add-on maintenance treatment for patients aged 12 years with severe eosinophilic asthma. Asthma is a chronic disease affecting more than 24 million people in the United States. It is characterized by periods of triggered exacerbations that cause airway inflammation, leading to difficulty in breathing and sometimes resulting in hospitalization.
FDA approval of mepolizumab was based on one dose-ranging trial and two confirmatory controlled trials in 1,327 patients. Study endpoints included frequency of exacerbations (trials 1 and 2), lung function as measured by volume of air exhaled in 1 second (all trials), and percentage of reduction of oral corticosteroid dose from baseline while maintaining asthma control (trial 3). Compared with placebo, patients receiving mepolizumab experienced significantly fewer exacerbations and were significantly more likely to reduce their daily maintenance oral corticosteroid dose by at least 50% while maintaining asthma control. However, mepolizumab treatment did not result in a significant improvement in lung function in any trial.
Pharmacology and Pharmacokinetics3,4: Mepolizumab is a humanized monoclonal antibody that binds to and inhibits interleukin-5 (IL-5), the primary cytokine responsible for eosinophil activation, recruitment, and differentiation. By inhibiting IL-5 signaling, mepolizumab effectively reduces the level of eosinophils in the blood, thereby limiting a key cellular component of the inflammatory process involved in asthma.
Mepolizumab is 80% bioavailable after SC injection, and its volume of distribution is estimated at 3.6 L. It is metabolized via proteolytic enzymes involved in endogenous protein catabolism throughout the body. Mepolizumab is cleared entirely metabolically at a rate of 0.28 L/day, and it has a mean terminal half-life of 16 to 22 days.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions reported in clinical trials of mepolizumab were headache, injection-site reactions, back pain, and fatigue. The use of mepolizumab was associated with an increased incidence of herpes zoster infection and possibly an increased risk of parasitic infections. As a monoclonal antibody, mepolizumab is associated with an increased risk of hypersensitivity reactions, and patients should be monitored after administration. Mepolizumab should not be used to treat acute bronchospasm or exacerbations, and patients receiving the drug should not have their oral or inhaled corticosteroid therapy reduced or discontinued abruptly. Data are insufficient regarding the safety of mepolizumab in pregnancy, but the drug may be transported across the placenta, and transport may increase as pregnancy progresses.
No formal drug-interaction studies involving mepolizumab have been performed, but the drug is not metabolized by CYP enzymes and it is not known to have any effect on CYP metabolism or P-glycoprotein pumps. Furthermore, there was no evidence of an effect of commonly coadministered drugs on mepolizumab exposure in population pharmacokinetic analyses during clinical trials.
Dosage and Administration3,4: Mepolizumab is supplied as 100-mg single-dose vials of lyophilized powder for reconstitution and SC injection by a healthcare professional. The recommended dosing is 100 mg by SC injection into the upper arm, thigh, or abdomen once every 4 weeks. The safety and efficacy of mepolizumab in patients aged <12 years have not been established. Although there are no trial data on patients with hepatic or renal impairment, no dose adjustments are specifically required, as it is unlikely that reduced hepatic or renal function would substantially affect the clearance rate of mepolizumab.
Secukinumab (Cosentyx, Novartis Pharmaceuticals)
Indication and Clinical Profile5,6: Secukinumab has been approved for the treatment of moderate-to-severe plaque psoriasis (Ps) in adults who are candidates for systemic therapy or phototherapy, and also for the treatment of adults with active psoriatic arthritis (PsA) or ankylosing spondylitis (AS). Ps, the most common form of psoriasis, is an autoimmune disorder of the skin characterized by the development of thick, red skin with flaky scales that cause significant skin irritation and discomfort. PsA is a form of inflammatory arthritis that develops in about 30% of people with psoriasis; the inflammation can affect the entire body and cause permanent joint and tissue damage. AS is a chronic disorder characterized by inflammation of the spine and the vertebral and sacroiliac joints.
FDA approval of secukinumab for Ps treatment was based on results of four multicenter, controlled trials in 2,403 adult Ps patients who were candidates for phototherapy or systemic therapy. Efficacy outcomes were the proportion of patients who achieved at least a 75% reduction in Psoriasis Area and Severity Index (PASI) score from baseline (PASI75) and treatment success (clear or mostly clear) on the Investigator’s Global Assessment (IGA-2011). In all studies, a significantly greater proportion of secukinumab patients versus placebo patients achieved both PASI75 (69% vs. 3%) and clear or mostly clear IGA (67% vs. 1.2%).
Approval of secukinumab for the treatment of PsA was based on results of two randomized, controlled studies (PsA1 and PsA2) involving 1,003 patients aged 18 years with active PsA despite nonsteroidal anti-inflammatory drug (NSAID), corticosteroid, or disease-modifying antirheumatic drug (DMARD) therapy. The primary endpoint was the percentage of patients achieving an Assessment in Ankylosing Spondylitis Response Criteria (ASAS20) response at week 24. In PsA1, patients treated with secukinumab 150 mg or 300 mg demonstrated a greater clinical response compared with placebo at week 24 (60% and 57% vs. 18%, respectively).
The efficacy of secukinumab in AS patients was assessed in two controlled studies (AS1 and AS2) involving 590 adult patients aged >18 years with active AS despite NSAID, corticosteroid, or DMARD therapy. The primary endpoint was the percentage of patients achieving an ASAS20 response at week 16. At week 16, patients treated with secukinumab 150 mg demonstrated greater improvement in ASAS20 response compared with those given placebo (61% vs. 28%, respectively).
Pharmacology and Pharmacokinetics5,6: Secukinumab is a human immunoglobulin G1 (IgG1) monoclonal antibody that binds selectively to the interleukin-17A (IL-17A) cytokine, blocking IL-17A from activating its receptor and effectively inhibiting the cytokine’s ability to trigger the inflammatory response.
The absolute bioavailability of secukinumab ranges from 55% to 77%. Secukinumab achieves peak serum concentration in approximately 6 days and reaches steady state by week 24 of regular dosing. It has a volume of distribution of 7 L to 8 L and is thought to be metabolized to small peptides via catabolic pathways analogous to endogenous IgG. The mean half-life of secukinumab is 22 to 31 days, and the drug is cleared at a rate of 0.14 L to 0.22 L per day.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions reported during clinical trials of secukinumab included nasopharyngitis, diarrhea, and upper respiratory tract infection. The drug carries warnings regarding an increased risk of infection, including tuberculosis (patients should be tested for tuberculosis prior to initiation); an increased risk of exacerbation in patients with Crohn’s disease; and a general warning regarding anaphylaxis. Secukinumab may be used during pregnancy only if the potential benefit justifies the potential harm to the fetus. It is not known whether secukinumab is excreted in breast milk.
Owing to the increased risk of infection, patients taking secukinumab should not receive live vaccinations. Although there is no reported role for IL-17A in the regulation of CYP enzymes, the formation of these enzymes is altered during chronic inflammation and therefore could be normalized by secukinumab. Because of this potential for interaction, monitoring is advised upon secukinumab initiation or discontinuation in patients who are currently taking other drugs that are CYP substrates and have a narrow therapeutic index.
Dosage and Administration5,6: Secukinumab is supplied as single-use, 150 mg/mL prefilled syringes or Sensoready pens and as 150 mg lyophilized powder in a single-use vial (for healthcare providers only). The recommended dosage for Ps is 300 mg by SC injection at weeks 0, 1, 2, 3, and 4 followed by 300 mg every 4 weeks. Each 300-mg dose is given as two injections of 150 mg. This regimen is recommended for PsA patients with coexistent moderate-to-severe Ps. In other PsA patients or those with AS, secukinumab may be administered with or without a loading dose (LD). The recommended dosage with an LD is 150 mg at weeks 0, 1, 2, 3, and 4 and every 4 weeks thereafter. Dosing without an LD is 150 mg every 4 weeks, possibly increasing to 300 mg if a patient continues to have active PsA. No dose adjustments are required in patients with renal or hepatic impairment or in elderly patients. Secukinumab has not been studied in pediatric patients.
Cariprazine (Vraylar, Allergan)
Indication and Clinical Profile7,8: Cariprazine is a new atypical antipsychotic approved for the treatment of schizophrenia and for the acute treatment of manic or mixed episodes associated with bipolar I disorder. Annually, schizophrenia affects about 1% of the U.S. population aged ≥18 years. The disorder is characterized by three symptom types: positive (abnormal behavior), negative (inability to perform daily activities), and cognitive (inability to make decisions). Patients with schizophrenia experience symptoms including multiple forms of hallucinations, paranoia, and social withdrawal. Bipolar disorder is a psychological condition characterized by alternating periods of mania and depression resulting in unusual shifts in mood, energy, and daily level of activity.
FDA approval of cariprazine for schizophrenia was based on results of three 6-week, randomized, placebo-controlled trials involving 1,754 patients with schizophrenia. The primary efficacy endpoint was symptom improvement from baseline measured by the Positive and Negative Syndrome Scale (PANSS). In all three trials, cariprazine over the dosage range of 1.5 to 9 mg per day was superior to placebo based on the PANSS total score (mean change –8.3 vs. placebo); however, there was a dose-related increase in certain adverse reactions, noted particularly at doses exceeding 6 mg.
FDA approval of cariprazine for manic or mixed episodes associated with bipolar I disorder was based on results of three 3-week placebo-controlled trials including 1,037 patients with bipolar I disorder with manic or mixed episodes with or without psychotic features. The primary efficacy endpoint was symptom improvement from baseline measured by the Young Mania Rating Scale (YMRS). In all three trials, cariprazine over the dosage range of 1.5 to 9 mg per day was superior to placebo based on the YMRS total score (mean change –5.6 vs. placebo); however, there was a dose-related increase in certain adverse reactions, noted particularly at doses exceeding 6 mg.
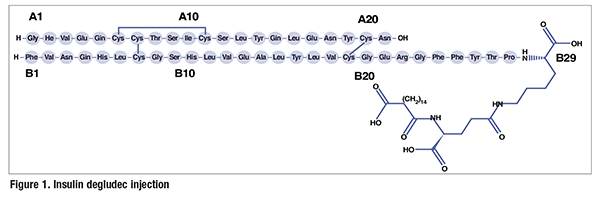
Pharmacology and Pharmacokinetics7,8: Cariprazine, which is a cyclohexylurea derivative of aripiprazole, acts as a partial agonist at central dopamine D₂ and serotonin 5-HT1A receptors, and it has antagonist activity at serotonin 5-HT2A receptors (FIGURE 2). Its exact mechanism of action in schizophrenia and bipolar I disorder is unknown, but is thought to be related to its ability as a partial agonist to antagonize dopamine receptors when dopamine levels are elevated (as is hypothesized in schizophrenic patients). Also, because of its activity as a partial agonist, this drug acts as an agonist when dopamine levels are low.
The bioavailability of cariprazine is 52%, and absorption is not affected by high-fat meals. Peak plasma concentrations are reached between 3 and 6 hours, and the drug is >90% bound by plasma proteins. Cariprazine undergoes extensive hepatic oxidation primarily by CYP3A4 and some CYP2D6, yielding the active metabolites desmethyl cariprazine and didesmethyl cariprazine (DDCAR). DDCAR undergoes further CYP3A4 hydroxylation to an inactive metabolite. The half-lives of cariprazine and DDCAR are 2 to 4 days and 1 to 3 weeks, respectively. Cariprazine is excreted primarily as metabolites.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions reported by patients receiving cariprazine in clinical trials included extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness. All atypical antipsychotics carry a black box warning alerting prescribers to an increased risk of death associated with off-label use to treat behavioral problems in older people with dementia-related psychosis. Like many atypical antipsychotics, cariprazine comes with warnings for cerebrovascular adverse effects, including stroke, neuroleptic malignant syndrome, tardive dyskinesia, metabolic changes, and orthostatic hypotension. Cariprazine should be used with caution in pregnant women because neonates exposed to the drug during the third trimester of pregnancy have experienced extrapyramidal and/or withdrawal symptoms.
Drug interactions with cariprazine are limited to those that are due to its metabolism by CYP3A4. If cariprazine is administered with a strong CYP3A4 inhibitor (e.g., clarithromycin, azole antifungals, protease inhibitors), the dose should be reduced to 75 mg. The use of cariprazine in patients taking a strong CYP3A4 inducer (e.g., rifampin, carbamazepine) should be avoided.
Dosage and Administration7,8: Cariprazine is supplied as 1.5-, 3-, 4.5-, and 6-mg capsules for oral administration once daily with or without food. The recommended starting dosage for both indications is 1.5 mg daily, which may be increased to 3 mg on day 2 of therapy. The recommended dosage range for schizophrenia and bipolar disorder is 1.5 to 6 mg per day and 3 to 6 mg per day, respectively, with a maximum recommended dosage of 6 mg. Safety and effectiveness have not been established in pediatric patients. No dose adjustments of cariprazine are required in patients with mild-to-moderate hepatic impairment (Child-Pugh score 5-9) or mild-to-moderate renal impairment (creatinine clearance ≥30 mL/min). Cariprazine should not be used in patients with severe renal or hepatic impairment, as the drug has not been adequately studied in this patient population.
Brivaracetam (Briviact, UCB)
Indication and Clinical Profile9,10: Brivaracetam was approved as an adjunctive therapy with other medications to treat partial-onset seizures in patients aged ≥16 years. Epilepsy, one of the most common central nervous system (CNS) pathologies, may be caused by stroke, infection, tumors, traumatic brain injury, or abnormal brain development. In many patients, the specific cause is not known. Approximately 5.1 million people in the United States have a history of epilepsy, and approximately 2.9 million have active epilepsy. Patients have different responses to the various seizure medications available, and brivaracetam offers patients a new treatment option.
FDA approval of brivaracetam was based on three phase III trials (studies 1-3). These multicenter, double-blind, controlled trials enrolled 1,550 patients with partial-onset seizures not adequately controlled with one or two concomitant antiepileptic therapies. All trials had an 8-week baseline period to identify patients who had at least eight partial-onset seizures. The baseline period was followed by a 12-week treatment period. Study 1 compared brivaracetam dosages of 50 mg and 100 mg per day with placebo; study 2 compared a dosage of 50 mg per day with placebo, and study 3 compared dosages of 100 mg and 200 mg per day with placebo. Compared with placebo, brivaracetam demonstrated statistically significant reductions (19.5%, 24.4%, and 24.0% for 50, 100, and 200 mg/day, respectively) in partial-onset seizure frequency per 28 days (P <.01). The proportion of patients with a ≥50% reduction in partial-onset seizure frequency was 34.2% (50 mg/day), 39.5% (100 mg/day), and 37.8% (200 mg/day), respectively, versus 20.3% for placebo (P <.01 for all arms).
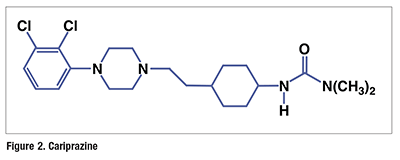
Pharmacology and Pharmacokinetics9,10: Brivaracetam is the 4-n-propyl derivative of the previously approved anticonvulsant levetiracetam (Keppra; FIGURE 3). The precise mechanism by which brivaracetam exerts its therapeutic activity is not known; however, the drug is bound with high affinity and selectivity by the synaptic vesicle protein 2A in various regions of the CNS, and this appears to contribute to the anticonvulsant effect. In animal models, brivaracetam is approximately 10 times more potent than levetiracetam in seizure prevention.
Brivaracetam is rapidly and completely absorbed after oral administration, providing peak levels within an hour. Administration with a high-fat meal slows the rate, but not the extent, of absorption. The drug is rapidly and evenly distributed to most tissues and is minimally bound by plasma proteins. Brivaracetam is metabolized primarily by amidase-mediated hydrolysis to the carboxylic acid and secondarily by CYP2C19 hydroxylation of the propyl side chain. Both metabolites are inactive. Therefore, to minimize brivaracetam overexposure and toxicity, dose reduction may be necessary in patients who are poor metabolizers owing to mutated CYP2C19 alleles, patients receiving concurrent CYP2C19 inhibitor therapy, and patients with hepatic disease. More than 95% of the dose is eliminated in the urine, primarily as metabolites. The terminal plasma half-life is about 9 hours.
Adverse Reactions and Drug Interactions9,10: The most common side effects associated with brivaracetam in clinical trials included drowsiness, dizziness, fatigue, nausea, and vomiting. This drug must be dispensed with a patient medication guide, which provides important information about brivaracetam’s use and risks. As is true of all drugs used to treat epilepsy, the most serious risks include suicidal thoughts, suicide attempts, feelings of agitation, new or worsening depression, aggression, and panic attacks. Rarely, patients may exhibit severe allergic reactions associated with swelling of the lips, eyelids, or tongue with or without difficulty breathing. Caution is advised in pregnancy because brivaracetam has been shown to cause fetal harm in animal models. Abrupt withdrawal of brivaracetam should be avoided in order to minimize the risk of increased seizure frequency and status epilepticus.
Brivaracetam does not produce clinically significant inhibition or induction of any of the primary CYP isozymes or major transporters in human tissues, and therefore it is unlikely to alter the metabolism or transport of other substrate drugs administered concurrently. Drugs that are CYP2C19 inducers (e.g., rifampin, phenytoin, and phenobarbital) can reduce plasma concentrations of brivaracetam by approximately 20% to 40%, and coadministration with rifampin may require a doubling of the brivaracetam dose. CYP2C19 inhibitors can cause modest plasma increases in brivaracetam plasma levels.
Dosage and Administration9,10: Brivaracetam is supplied in three formulations: tablet (10, 25, 50, 75, and 100 mg), oral solution (10 mg/mL), and injection for IV administration. The recommended oral starting dosage is 50 mg twice daily (100 mg/day). Based on individual patient tolerability and therapeutic response, the dosage may be adjusted down to 25 mg twice daily (50 mg/day) or up to 100 mg twice daily (200 mg/day). For all stages of hepatic impairment, the recommended starting dosage is 25 mg twice daily, with a maximum dosage of 75 mg twice daily. When oral dosing is temporarily not feasible, brivaracetam may be given parenterally in the same dosages employed for oral administration. When a patient discontinues brivaracetam, the drug should be withdrawn gradually.
REFERENCES
1. Tresiba (insulin degludec injection) package insert. Plainsboro, NJ: Novo Nordisk; September 2015.
2. Garber AJ, King AB, Del Prato S, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379:1498-1507.
3. Nucala (mepolizumab) package insert. Philadelphia, PA: GlaxoSmithKline; November 2015.
4. Bel EH, Wenzel SE, Thompson PJ, et al; SIRIUS Investigators. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189-1197.
5. Cosentyx (secukinumab) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; January 2016.
6. Langley RG, Elewski BE, Lebwohl MR, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. New Engl J Med. 2014;371:326-338.
7. Vraylar (cariprazine) package insert. Parsippany, NJ: Actavis Pharma, Inc; September 2015.
8. Gründer G. Cariprazine, an orally active D2/D3 receptor antagonist, for the potential treatment of schizophrenia, bipolar mania and depression. Curr Opin Investig Drugs. 2010;11:823-832.
9. Briviact (brivaracetam) package insert. Smyrna, GA: UCB, Inc; February 2016.
10. Malawska B, Kulig K. Brivaracetam UCB. Curr Opin Investig Drugs. 2005;6:740-746.
To comment on this article, contact rdavidson@uspharmacist.com.



