US Pharm. 2023;48(2):HS-2-HS-5.
ABSTRACT: Pulmonary hypertension is a rare, life-threatening progressive disease of the lungs characterized by increased pulmonary vascular resistance and narrowing of the blood vessels within the pulmonary vasculature. Patients with pulmonary hypertension often present with nonspecific symptoms such as fatigue, exertional dyspnea, lower-extremity edema, and syncope. Pulmonary hypertension can be classified into five broad categories that help direct treatment plans. Current therapies are primarily aimed at reducing functional impairment and treating the underlying conditions. Pharmacists must remain informed on the latest treatments and therapeutic approaches to contribute to the optimal management of patients with pulmonary hypertension.
Pulmonary hypertension (PH) is a rare, life-threatening progressive disease of the lungs characterized by increased pulmonary vascular resistance and narrowing of the blood vessels within the pulmonary vasculature.1 An estimated 10.6 cases per 1 million adults in the United States have been reported.2 PH can affect all age groups (including children), races, and sexes. PH incidence increases with age. It is more common in females, non-Hispanic black patients, and those older than age 75 years.3
PH is defined by a mean pulmonary arterial pressure (mPAP) >20 mmHg at rest as assessed by a right heart catheterization.4,5 Evidence has shown the normal mPAP is between 14 +/- 3 mmHg and 20 mmHg.6 This definition recently changed from previous guideline iterations, which defined PH as mPAP ≥25 mmHg at rest. This change occurred since evidence suggests that patients with a mPAP of 21 mmHg to 24 mmHg are at a higher risk of mortality and hospitalizations.4,5,7,8
Presenting signs and symptoms of PH are usually subtle and nonspecific, and they progressively worsen over time.8 Upon examination, providers may hear a loud second heart sound at disease onset. If a right-sided third heart sound, tricuspid regurgitation murmur, increased jugular venous distension, or a right ventricular heave is noted, the patient may have more advanced PH.9 With regard to symptoms, patients most commonly first complain of dyspnea followed then by fatigue, syncope, palpitations, angina, and weight gain. As a result of the subtle presentation of PH, diagnosis is often delayed until symptoms deteriorate.9,10 As such, pulmonary function tests, cardiac echocardiography, and an assessment for myocardial ischemia are recommended to help confirm the diagnosis of PH.11,12
While most cases of PH are idiopathic, PH is associated with a variety of clinical conditions.4 PH is classified into the following five categories based on etiology: 1) pulmonary arterial hypertension (PAH); 2) PH associated with left heart disease; 3) PH associated with lung diseases and/or hypoxia; 4) PH associated with chronic pulmonary artery obstruction; and 5) PH with unclear and/or multifactorial mechanisms.4 The classification of the PH, severity of symptoms, and/or underlying etiology can help determine the appropriate treatment plan. Current therapies are primarily aimed at reducing functional impairment and mortality risk by increasing exercise capacity, improving quality of life, and preserving right ventricular function.13 The purpose of this article is to provide pharmacists a brief overview of the latest guideline-based treatment recommendations and therapeutic approaches in PH.
Treatment
Since there is no cure for PH, current medications and procedures attempt to slow the progression of the disease and improve a patient’s quality of life. Currently, there are three main medication classes to manage PH. Each class targets a separate pathophysiologic pathway but primarily works by causing and/or targeting pulmonary artery vasodilation. For instance, endothelin receptor antagonists (ambrisentan, bosentan, and macitentan) work by antagonizing the endothelin system, leading to decreased levels of the vasoconstrictor endothelin-1 in PH patients.14 Alternatively, phosphodiesterase-5 (PDE-5) inhibitors (sildenafil, tadalafil, vardenafil) and guanylate cyclase stimulators (riociguat) work by preventing the degradation of cyclic guanosine monophosphate (cGMP). cGMP causes vasodilation through the nitric oxide/cGMP pathway, and PDE-5 is responsible for degrading cGMP.15 The third class of medications is the prostacyclin analogues (beraprost, epoprostenol, iloprost, treprostinil) and prostacyclin receptor agonists (selexipag), which work by enhancing prostacyclin.16 Prostacyclin is a potent vasodilator, and dysregulation of prostacyclin synthesis metabolism has been identified in PH patients. Additional supportive therapies that should be considered for PH patients include oxygen supplementation, diuretics, digoxin, exercise, and anticoagulation.
Group 1: Pulmonary Arterial Hypertension
PAH, a subtype of PH, can be further classified as idiopathic, heritable, or drug/toxin–induced and associated with known conditions that cause lesions in the small pulmonary arterioles (e.g., HIV infection, portal hypertension).4 The World Health Organization (WHO) further categorizes PAH into functional classes denoting severity of the disease (TABLE 1).17
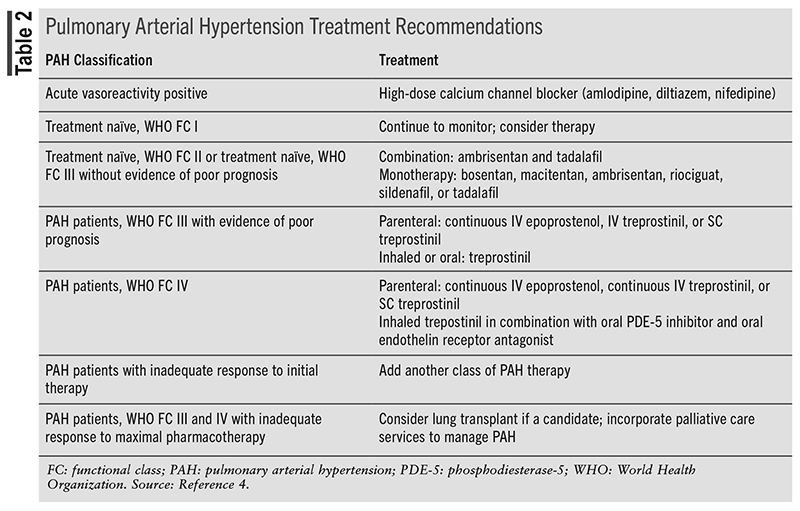
Current guidelines recommend that selection of PAH-specific therapy be based on WHO functional classes.4 PAH-specific agents include prostacyclin pathway agonists, endothelin receptor antagonists, nitric oxide-cGMP enhancers, or, rarely, calcium channel blockers (TABLE 2).4 Calcium channel blocker therapy can be considered in patients with idiopathic PAH, heritable PAH, and drug/toxin–induced PAH who who have a positive acute vasoreactivity test.4 Current literature does not recommend specific agents over others when there are multiple options for monotherapy.4 Combination therapy can carry a more intense adverse reaction profile that patients may not want to endure, and having multiple medications can be a financial burden. Additionally, patient-specific factors guide clinical decisions, whether it is patient preference, adverse reactions, cost, or insurance coverage.4 Following initiation of therapy, patients should follow up with providers within the first 6 weeks and every 3 months thereafter. Despite treatment advances, the 5-year survival rate following PAH diagnosis has been reported to be 43.8%.18

Group 2: Pulmonary Hypertension Secondary to Left Heart Disease
Group 2 PH is the most common form of PH, representing 65% to 80% of all cases.4,19,20 Group 2 PH is characterized by left heart disease commonly due to heart failure (HF), including HF with reduced ejection fraction, HF with mid-range ejection fraction, HF with preserved ejection fraction, and HF caused by left-sided valvular disease.20 As such, therapy for Group 2 PH focuses on treatment of the cardiovascular diseases causing PH and optimizing current HF therapies to maintain the right ventricular function.4,21 PAH-specific therapies are not recommended with Group 2 PH due to limited supportive evidence of clinical benefit and increased risk of mortality.22,23 Referral to specialized clinics for treatment on an individual basis is ideal for Group 2 PH management.22
Group 3: Pulmonary Hypertension Secondary to Lung Disease
PH patients with chronic obstructive pulmonary disease, emphysema, interstitial lung disease (ILD), combined pulmonary fibrosis and emphysema, or hypoventilation syndromes are categorized as having Group 3 PH.4 The goal of therapy for patients in this PH subgroup is to optimize therapy of the underlying lung disease. For most patients classified as having Group 3 PH, PAH-specific therapy is not recommended since there is conflicting evidence due to varying outcomes and the potential for detrimental hemodynamic effects.4 Inhaled treprostinil, a prostacyclin analogue, was approved in May 2022 by the FDA for the treatment of PAH (WHO Group 1) and PH associated with ILD (PH-ILD; WHO Group 3) to improve exercise ability. It is the first and only FDA-approved agent for use in PAH and PH-ILD. FDA approval was supported by data from the BREEZE and INCREASE studies, which demonstrated improved exercise capacity and quality of life in patients in these PH subgroups.24,25 Oxygen or noninvasive ventilation, smoking cessation counseling, and exercise should be considered adjunctive therapies.4
Group 4: Pulmonary Artery Obstruction
PH due to pulmonary artery obstruction, otherwise known as chronic thromboembolic pulmonary hypertension (CTEPH), is a complication of an acute pulmonary embolism.4,26,27 The primary treatment option for these patients is to undergo a pulmonary endarterectomy (PEA) to remove the obstruction.4,28 Prior to the PEA, patients may receive an endothelin receptor antagonist or a PDE-5 inhibitor.29,30 After undergoing PEA, patients must start reocclusion prophylaxis with heparin followed by oral anticoagulation (warfarin or direct-acting oral anticoagulant) indefinitely to prevent further thromboembolisms.29 Around 40% of the patients enrolled in the CTEPH registry were deemed inoperable and were recommended medication management.31
The only FDA-approved treatment for patients who are deemed inoperable is the drug riociguat.32 Riociguat is a soluble guanylate cyclase stimulator that works by promoting the nitric oxide pathway.32 Riociguat increases nitric oxide, increasing cGMP levels and resulting in vasodilation. Riociguat should be initiated at 1 mg orally three times a day.33 The dose can be started at half the recommended dose (0.5 mg) if there is risk for hypotension.33 Titration by 0.5 mg can occur in 2-week periods until maximum dosage of 2.5 mg three times a day is reached.33 One important consideration when using this medication is the black box warning for pregnant patients, as it may lead to embryo-fetal toxicity.33
Group 5: Unclear or Multifactorial Causes
Patients with PH due to unclear or multifactorial causes can be a challenge to treat due to a poor understanding of the correlation between PH and the disorder. Therefore, treatment recommendations vary, and additional research is needed to identify a correlation between these disorders and PH to make strong recommendations using PH-specific agents. When treating these disorders, it is advised to treat the condition while considering the treatment of PH related to it.4 There are some noteworthy disorders, such as chronic myelogenous leukemia, sarcoidosis, pulmonary Langerhans cell histiocytosis, and Gaucher disease, with specific treatments that also benefit PH.4,34-36 However, most of the data to support the use of these medications are based on small research studies and are less specific in nature, so caution is advised in using them to recommend therapy.
The Pharmacist’s Role
Optimal care for patients with PH should be provided by a collaborative, interdisciplinary clinical team. Because of the complexities and intricacies of PH drug therapy, pharmacists can play a key role. Prior to selection, pharmacists can ensure that the patient will have access to the drug since most PH medications are costly. Some of the approved PH drug therapies have specific administration methods or dosing strategies. Pharmacists can develop standardized medication order sets that lower the chance for a medication error. Following selection and initiation of drug therapy, concomitantly administered medications should be reviewed by pharmacists for any potential drug interactions. Pharmacists can provide ways to mitigate or avoid interactions. Lastly, pharmacists must remain informed on all of the nuances of these complex regimens. Remaining up to date can better equip pharmacists to provide medication education to healthcare workers, patients, and caregivers.
Conclusion
PH is a severe disorder of the pulmonary vasculature, usually secondary to a variety of cardiac and pulmonary conditions. Diagnosis is challenging and delayed as patients commonly present with nonspecific symptoms. Monotherapy or combination drug therapy targeting multiple pathways can be used to improve outcomes for patients with PH. However, if left untreated, PH can progress to right HF and eventually death. PH is best managed by an interprofessional team that includes pharmacists to better enhance appropriate and safe medication use.
REFERENCES
1. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376-387.
2. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national perspective registry. Ann Intern Med. 1991;115(5):343-349.
3. George MG, Schieb L, Ayala C, et al. Pulmonary hypertension surveillance: United States, 2001 to 2010. Chest. 2014;146(2):476-495.
4. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-3731.
5. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913.
6. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888-894.
7. Simonneau G, Hoeper MM. The revised definition of pulmonary hypertension: exploring the impact on patient management. Eur Heart J Suppl. 2019;21(Suppl K):K4-K8.
8. Maron BA, Hess E, Maddox TM, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veterans Affairs Clinical Assessment, Reporting, and Tracking Program. Circulation. 2016;133(13):1240-1248.
9. Braganza M, Shaw J, Solverson K, et al. A prospective evaluation of the diagnostic accuracy of the physical examination for pulmonary hypertension. Chest. 2019;155(5):982-990.
10. Brown LM, Chen H, Halpern S, et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL registry. Chest. 2011;140(1):19-26.
11. Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial hypertension: a review [published correction appears in JAMA. 2022;328(9):892]. JAMA. 2022;327(14):1379-1391.
12. Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53(1):1801904.
13. McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D73-D81.
14. Correale M, Ferraretti A, Monaco I, et al. Endothelin-receptor antagonists in the management of pulmonary arterial hypertension: where do we stand? Vasc Health Risk Manag. 2018;14:253-264.
15. Unegbu C, Noje C, Coulson JD, et al. Pulmonary hypertension therapy and a systematic review of efficacy and safety of PDE-5 inhibitors. Pediatrics. 2017;139(3):e20161450.
16. Lang IM, Gaine SP. Recent advances in targeting the prostacyclin pathway in pulmonary arterial hypertension [published correction appears in Eur Respir Rev. 2016 Mar;25(139):99] [published correction appears in Eur Respir Rev. 2017 Jan 3;26(143):155067]. Eur Respir Rev. 2015;24(138):630-641.
17. Highland KB, Crawford R, Classi P, et al. Development of the Pulmonary Hypertension Functional Classification Self-Report: a patient version adapted from the World Health Organization Functional Classification measure. Health Qual Life Outcomes. 2021;19(1):202.
18. Farber HW, Miller DP, Poms AD, et al. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148(4):1043-1054.
19. Weitsman T, Weisz G, Farkash R, et al. Pulmonary hypertension with left heart disease: prevalence, temporal shifts in etiologies and outcome. Am J Med. 2017;130(11):1272-1279.
20. Clark CB, Horn EM. Group 2 pulmonary hypertension: pulmonary venous hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(3):401-411.
21. Al-Omary MS, Sugito S, Boyle AJ, et al. Pulmonary hypertension due to left heart disease: diagnosis, pathophysiology, and therapy. Hypertension. 2020;75(6):1397-1408.
22. Desai A, Desouza SA. Treatment of pulmonary hypertension with left heart disease: a concise review. Vasc Health Risk Manag. 2017;13:415-420.
23. Vachiéry JL, Tedford RJ, Rosenkranz S, et al. Pulmonary hypertension due to left heart disease. Eur Respir J. 2019;53(1):1801897.
24. Spikes LA, Bajwa AA, Burger CD, et al. BREEZE: Open-label clinical study to evaluate the safety and tolerability of treprostinil inhalation powder as Tyvaso DPI™ in patients with pulmonary arterial hypertension. Pulm Circ. 2022;12(2):e12063.
25. Nathan SD, Tapson VF, Elwing J, et al. Efficacy of inhaled treprostinil on multiple disease progression events in patients with pulmonary hypertension due to parenchymal lung disease in the INCREASE trial. Am J Respir Crit Care Med. 2022;205(2):198-207.
26. Delcroix M, Torbicki A, Gopalan D, et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur Respir J. 2021;57(6):2002828.
27. Kim NH, Delcroix M, Jais X, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(1):1801915.
|28. Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S67-S77.
29. Mayer E. Surgical and post-operative treatment of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2010;19(115):64-67.|
30. Condliffe R, Kiely DG, Gibbs JS, et al. Improved outcomes in medically and surgically treated chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. 2008;177(10):1122-1127.
31. Logue R, Safdar Z. Chronic thromboembolic pulmonary hypertension medical management. Methodist Debakey Cardiovasc J. 2021;17(2):e29-e33.
32. Khaybullina D, Patel A, Zerilli T. Riociguat (adempas): a novel agent for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. P T. 2014;39(11):749-758.
33. Adempas (riociguat) [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2017.
|34. Adir Y, Humbert M. Pulmonary hypertension in patients with chronic myeloproliferative disorders. Eur Respir J. 2010;35(6):1396-1406.
35. Baughman RP, Shlobin OA, Gupta R, et al. Riociguat for sarcoidosis-associated pulmonary hypertension: results of a 1-year double-blind, placebo-controlled trial. Chest. 2022;161(2):448-457.
36. Le Pavec J, Lorillon G, Jaïs X, et al. Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest. 2012;142(5):1150-1157.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



