US Pharm. 2021;46(3):HS1-HS10.
ABSTRACT: Cancer-induced pain reflects a multiplicity of underlying mechanisms. It may arise from nociceptive damage, neuropathic damage, or both, and neuropathic pain may also be related to treatment. Initial and ongoing assessment of pain should be an integral part of supportive oncology. A variety of options are available for pain management, depending on the type and source of the pain. Opioids are the mainstay of management of moderate-to-severe cancer-induced pain. Interventional management of pain, which comprises a number of invasive analgesic therapies, may be used in selected patients. Buprenorphine, a biased opioid agonist, produces analgesia with fewer adverse effects. The use of pharmacogenomic testing for opioids remains controversial in cancer patients.
The International Association for the Study of Pain has defined pain as “an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage.”1 This definition was expanded in July 2020 to include the physical, psychological, and social contexts of pain.1 It has been estimated that 35% to 96% of cancer patients are affected by pain.2 Pain has a high prevalence in certain types of tumors, such as pancreatic adenocarcinoma and head and neck cancer.3 Poorly controlled pain can have devastating effects on a patient’s quality of life and physical functioning, resulting in increased distress. About 90% of patients can achieve good pain control by following established guidelines.4 Safe, effective, and evidence- or consensus-based guidelines on the management of cancer-related pain are a cornerstone of comprehensive cancer care.5-7
Despite an increased interest in—and efforts to improve—the management of cancer-related pain, this area is often overlooked or remains suboptimal owing to limited time in the ambulatory-care setting, failure to refer patients to appropriate providers to manage pain adequately and promptly, and underestimation of a patient’s pain. This article presents an overview of therapeutic advances in cancer-related pain management.
PATHOPHYSIOLOGY OF CANCER-INDUCED PAIN
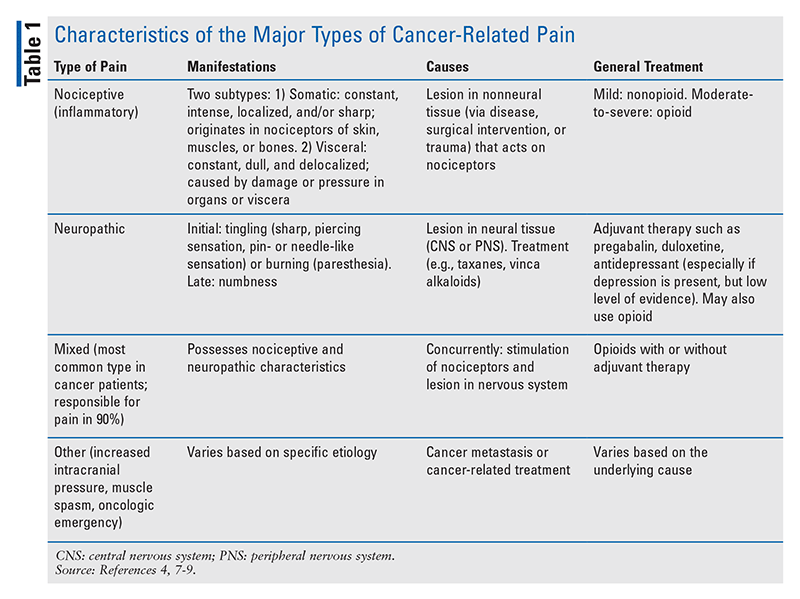
Cancer-induced pain is not homogeneous; it reflects a multiplicity of underlying mechanisms. It may arise from nociceptive damage, neuropathic damage, or both (i.e., mixed pain).8 Neuropathic pain may also be related to treatment. In general, nociceptive or inflammatory pain arises from stimuli within damaged tissue (e.g., disease, surgery, trauma), whereas neuropathic pain is caused by direct lesion of the sensory nerves (e.g., diabetic neuropathy, herpetic neuralgia, radiculopathy). Nociceptive pain can be further categorized as visceral (internal organs) or somatic (skin, muscle, bone). Cancer-induced pain may be mixed because while, on the one hand, tumor growth induces tissue damage and the release of various inflammatory mediators, the compression, infiltration, or obstruction of tissue and injury to nerve fibers result in neuropathic pain.8 Notably, adverse effects (AEs) associated with systemic chemotherapy (e.g., oxaliplatin- or taxane-induced peripheral neuropathy), radiation (e.g., mucositis, dermatitis), and aromatase inhibitors (e.g., myalgia, arthralgia) also contribute to increased nociception.4 TABLE 1 summarizes the characteristics of the major types of cancer-related pain.4,7-9
The presence and intensity of cancer-related pain do not correlate with the size or number of active malignancies.8 Cancer-induced damage to the sensory nerves (peripheral nervous system) leads to transmission of altered impulses to the sensory neurons in the dorsal horn of the spinal cord (central nervous system), resulting in a state of general hyperexcitability.9 It is now widely recognized that pain can be modulated by multiple factors, such as mood, chemical coping, and cognition; therefore, it is important to identify these modulating factors in pain management.4 For example, an abdominal-pain score of 9 out of 10 may be due to visceral pain in one cancer patient but in another patient may be due to hyperalgesia.4 Accordingly, an opioid-dose increase would be warranted in the first patient, whereas in the second patient either the medication should be switched to a different opioid or the delirium should be treated.4
CLINICAL ASSESSMENT
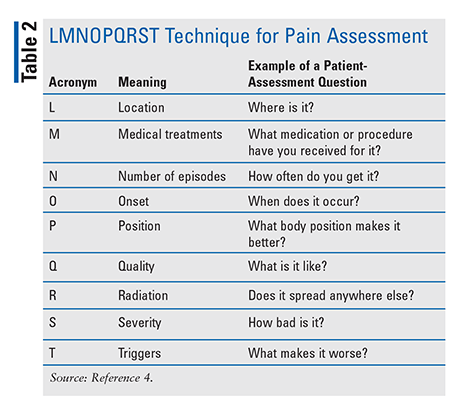
Chronic cancer pain is among the most significant of all cancer symptoms in terms of prevalence and potential consequences, and it is essential to follow best practice (humane, effective, and affordable care) in the management of cancer pain. Many tools are available to assess pain, including the Visual Analogue Scale, the Wong-Baker FACES pain rating scale, and the Iowa Pain Thermometer.10-12 Pain can also be assessed by a numerical rating system (e.g., a scale of 0-10 where 0 = no pain and 10 = worst pain possible) or a verbal-descriptor scale (e.g., pain is described as none, mild, moderate, or severe). A mnemonic for assessing cancer pain—LMNOPQRST—is a useful tool for making a diagnosis as well as personalizing treatment (TABLE 2).4 For patients with severe cognitive impairment, facial expression and changes in body movement may be reliable indicators of the presence of pain but not the pain’s intensity.5,13
Pain related to an oncologic emergency (i.e., life-threatening event related to a patient’s malignancy) may be due to pathologic fracture, leptomeningeal metastasis, infection, or an obstructed or perforated viscus.7 Pain associated with an oncologic emergency should be addressed immediately via concurrent treatment of the underlying condition.7
RECENT ADVANCES IN PHARMACOLOGIC MANAGEMENT
Opioid receptors (ORs), which are members of the G protein–coupled receptor (GPCR) superfamily, mediate nociceptive, hedonic, emotional, autonomic, neuroendocrine, and immune responses.14,15 There are four OR subtypes: mu-OR, kappa-OR, delta-OR, and OR-like 1 receptor. mu-OR represents the primary target for conventional opioids such as morphine. The development of opioid tolerance and dependence results from excessive mu-OR activation throughout the nervous system.16 kappa-OR mediates dysphoria and opposes mu-OR in regulating the hedonic tone, whereas delta-OR regulates emotional responses and reduces hyperalgesia.16,17
Full or pure opioid agonists such as fentanyl and morphine induce the maximal signaling response, whereas partial agonists (e.g., buprenorphine) and antagonists (e.g., naloxone) promote submaximal signaling and decrease basal activity, respectively.17 Buprenorphine is also a biased agonist that selectively activates certain receptor-associated pathways at the expense of others.18 Although mu-OR, kappa-OR, and delta-OR inhibit nociceptive responses, each has a specific role in regulating the wide spectrum of pain modalities.
A second set of intracellular proteins, called beta-arrestins, are now known to regulate mu-OR functions and play an important role in modulating mu-OR signaling and pharmacology.19 As mentioned, conventional opioids bind to the mu-OR and produce analgesia, mainly via the GPCR pathway. At the same time, these opioids trigger the beta-arrestin pathway, which is responsible for AEs such as nausea, vomiting, constipation, respiratory depression, tolerance, and dependence.16 On the other hand, biased agonists such as buprenorphine preferentially activate the GPCR over the beta-arrestin pathway, thereby producing analgesia with diminished AEs.20
Current therapeutic strategies for the development of novel analgesics with fewer AEs focus on either modulating the activities of specific receptors or deploying biased agonists. Compared with conventional opioids, buprenorphine dissociates more slowly from the mu-OR, resulting in prolonged analgesia and less potential for withdrawal.21,22 Because it also blocks delta-OR and kappa-OR, making them less likely to induce sedation, dysphoria, constipation, and hyperalgesia, buprenorphine represents a novel agent in cancer-pain management. Oliceridine (Olinvyk), a full mu-OR agonist available as an IV formulation that received regulatory approval for acute pain in 2020, is another biased agonist that can reduce AEs of conventional opioids.23
SAFE PRESCRIBING OF OPIOIDS
In 2016, in recognition of the rising death rate from opioids and with the goal of stemming opioid abuse, the CDC issued a guideline recommending opioid use at the lowest appropriate dosage for the shortest period of time for chronic noncancer pain.24 Although the CDC guideline was addressing noncancer providers, it led to unnecessary fear about prescribing opioids even for cancer-induced pain at appropriate dosages. Subsequently, the American Society of Clinical Oncology released a guideline advocating the judicious use of opioids in patients who do not respond to conservative pain management; this guideline included a precaution to minimize opioid abuse and addiction.25 International guidelines and studies support the idea that opioids should be continued in advanced malignancies for cancer-induced pain.5,7,26 Most importantly, there is no justification for stopping or reducing appropriately titrated opioids for end-of-life care.26 Validated assessment tools to screen for and monitor aberrant opioid use are available.7,27
INDIVIDUALIZED PAIN-MANAGEMENT CONSIDERATIONS
Choice of Pharmacologic Agent
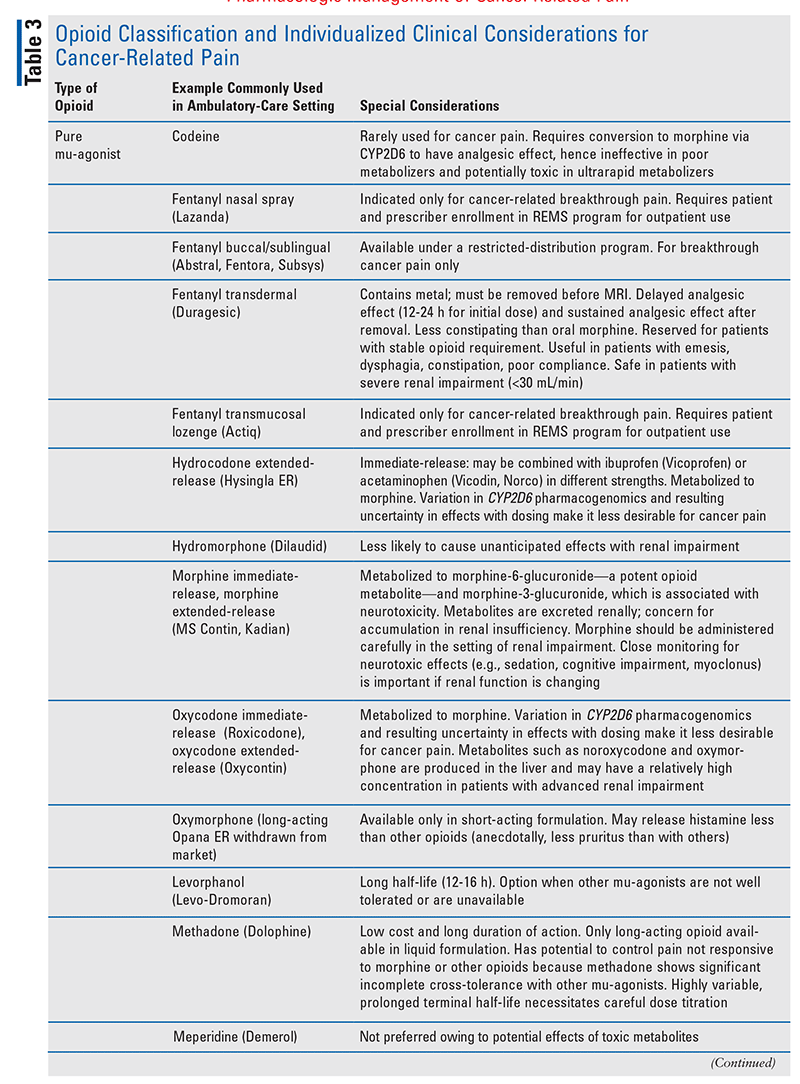
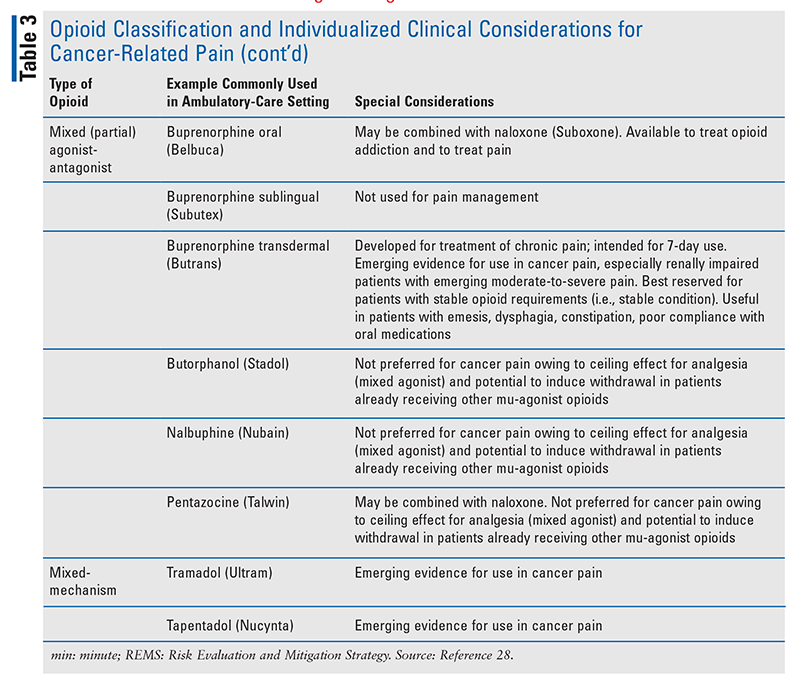
Opioids continue to be the mainstay of treatment for cancer-related pain, along with adjuvants (also known as adjunctive treatments or coanalgesics) to control neuropathic pain. Each opioid is associated with a specific AE profile, necessitating individualized consideration for clinical use, including in the ambulatory-care setting (TABLE 3).28 For example, the use of morphine in patients with renal failure can cause an increase in morphine metabolites, leading to neurotoxicity and increased risk of accidental overdose.29 By contrast, because oxycodone is metabolized in the liver, hepatic failure or cachexia may increase the medication’s half-life.
Transdermal fentanyl has a delayed onset of action when first applied, and the patient must be monitored for reservoir effect when the patch is removed and a new opioid is added to the patient’s regimen. It may be used in patients who are opioid-tolerant, are converted from a continuous IV opioid, or have difficulty swallowing oral opioids. Transdermal fentanyl may have decreased absorption in patients with cachexia, so the risk of potential high dose must be carefully monitored.29
Buprenorphine, which is available in both transdermal (Butrans) and sublingual (Zubsolv) formulations, is a partial mu-OR agonist that is used for opioid-replacement therapy in patients with opioid addiction. It exhibits a ceiling to analgesic efficacy and therefore may precipitate withdrawal effects in patients taking high-dose opioids.7 Buprenorphine may be a first-line agent for chronic cancer pain in both opioid-naïve and opioid-tolerant patients because it causes fewer AEs and is better tolerated in elderly patients compared with other opioids. It may be used in patients with end-stage renal disease.7
Methadone is a mu-OR agonist as well as an N-methyl-d-aspartate receptor antagonist. Because of its high potency, long half-life, and significant interindividual variation in pharmacokinetics, methadone requires careful titration.7 Recent evidence suggests that appropriately titrated methadone has similar efficacy and tolerability to morphine for managing cancer-related pain.30
Adjuvants include tricyclic antidepressants (TCAs) such as amitriptyline and nortriptyline, serotonin-norepinephrine reuptake inhibitors such as duloxetine and venlafaxine, anticonvulsants such as gabapentin and pregabalin, topical anesthetics (e.g., lidocaine patch), topical therapies (e.g., capsaicin), corticosteroids, bisphosphonates, and cannabinoids.31 Nonopioid analgesics include nonsteroidal anti-inflammatory drugs and acetaminophen. TCAs are useful in cancer patients experiencing depression or insomnia. Gabapentin, pregabalin, duloxetine, and TCAs are strongly recommended as single agents for first-line neuropathy treatment.5 Adjuvants may be added to opioids; however, pain reduction greater than 1 point on a numerical rating scale of 1 to 10 probably will not be achieved with combination therapy, and the probability of AEs will likely increase.32
Interventional Pain Management
Interventional pain management comprises a number of invasive analgesic therapies, including injection-based treatments, catheter-based infusion therapies, implanted devices, and some surgical approaches.33,34 These therapies include percutaneous or neuraxial (epidural or intrathecal) administration of pain medications such as opioids, baclofen, clonidine, steroids, and local anesthetics. Injection of a bone cement (e.g., polymethylmethacrylate or its alternative) into the compressed vertebral body, known as vertebroplasty or kyphoplasty, is a minimally invasive technique that helps manage pain from pathologic fractures caused by osteoporosis, metastasis, or multiple myeloma.7,33 The evidence for the efficacy of these interventional strategies remains largely anecdotal and poorly understood, especially in cancer patients, but these techniques may be safe and effective in carefully selected individuals.34
Compared with somatic pain, cancer pain of visceral origin is often vague and poorly localized.35 Afferent nerve fibers that mediate pain innervate the paravertebral sympathetic ganglia. Sympathetic nerve block, which interrupts the action of these visceral afferent fibers, is an effective means of controlling severe visceral cancer pain.35 Common sites for sympathetic nerve block include the stellate ganglion (to interrupt pain in head, neck, upper extremities, and intrathoracic structures), lumbar sympathetic trunk (to interrupt pain in lower extremities), celiac plexus, superior hypogastric plexus, and ganglion impar. The last three of these sites collectively block visceral abdominal and pelvic pain. Sympathetic nerve fibers in these sites are anatomically demarcated from somatic nerves, thus sparing sensory and motor fibers during the procedure.34
By contrast, somatic nerve block involves interrupting both sensory and motor neural activities.34 It can be useful for localizing the afferent pathway involved in sustaining the pain, such as somatic pain arising from the abdominal wall versus referred pain from a visceral organ.34 Both sympathetic and somatic nerve blocks can be neurolytic or nonneurolytic. Nonneurolytic block involves the use of a local anesthetic (e.g., lidocaine, bupivacaine) to provide temporary relief and help map the location of somatic or visceral pain. Neurolytic block is the intentional destruction of nervous tissue with phenol or alcohol to achieve pain relief. Sedation using higher doses of opioids may be indicated in opioid-tolerant cancer patients receiving nerve blocks. It should be noted that neurolytic block of somatic nerves may be complicated by postneurolysis neuritic pain. This concern often restricts the use of neurolytic block to those patients with advanced malignancies who have a limited life expectancy.7,35
Drug-Drug Interactions
If adjuvant medications such as TCAs are prescribed, the risk of serotonin syndrome with selective serotonin reuptake inhibitors (SSRIs) must be carefully evaluated. The rising use of opioids and uptick in prescriptions for serotonergic drugs have also increased the risk of serotonin syndrome.36 Although clinical data are limited, several cases of opioid-associated serotonin toxicity have been reported.37,38 An association between opioids and either an SSRI or a monoamine oxidase inhibitor was found in more than 25% of cases of serotonin syndrome in a retrospective analysis.39 Despite the fact that case reports and retrospective reviews generally represent a low level of evidence, proper education of prescribers and patients and early recognition of signs and symptoms of this potential drug-induced AE are important.
Some antidepressants may inhibit the CYP450 isoenzymes, particularly 2D6. Tamoxifen, a selective estrogen receptor modulator commonly used in patients with endocrine receptor–positive breast cancer, is metabolized by 2D6 into active metabolites. The inhibition of CYP2D6 may decrease the efficacy of tamoxifen. However, it remains controversial whether tamoxifen-treated patients who also received SSRIs have an increased risk of breast cancer recurrence versus those patients on tamoxifen alone.40-42 Current National Comprehensive Cancer Network guidelines recommend that if concomitant use of tamoxifen and SSRI is required, a mild CYP2D6 inhibitor (e.g., citalopram, escitalopram, sertraline, venlafaxine) may be preferred over a moderate-to-potent inhibitor (e.g., bupropion, duloxetine, fluoxetine, fluvoxamine, paroxetine).7
Equianalgesic Conversion of Opioids
Reasons for changing from one or more opioids to a different opioid include lack of efficacy, development of tolerance or intolerable AEs, and hyperalgesia.43,44 Equianalgesic opioid-conversion tables and online opioid-conversion calculators are readily available to aid clinicians in dose conversion, but many opioid-conversion formulas vary in their conversion methodology. To date, there is no evidence that one conversion ratio is superior to others. In fact, equianalgesic opioid conversion should not be a pure mathematical calculation but rather must be part of a comprehensive assessment of opioid therapy, including LMNOPQRST.4,45
In addition, when making an opioid conversion, it is important to reduce the dose of the new opioid by 10% to 25% to account for incomplete cross-tolerance.7 Incomplete cross-tolerance may develop because tolerance to one opioid does not imply complete tolerance to another during conversion. Opioid conversion or rotation without taking this concept into account may lead to accidental sedation or unintended overdose.45
Pharmacogenomic Testing of Opioids
Opioids are metabolized mainly by CYP isoenzymes or uridine diphosphate-glucuronosyl-transferase in the liver. The CYP2D6 isoenzyme metabolizes codeine, hydrocodone, oxycodone, and tramadol to the active metabolites morphine, hydromorphone, oxymorphone, and O-desmethyltramadol, respectively. CYP2D6 gene polymorphism alters enzyme activity and results in the generation of four variant isoenzymes with varied metabolism of some medications (including opioids), namely, ultrarapid metabolizer, extensive or normal metabolizer, intermediate metabolizer, and poor metabolizer.46 In a study of patients with noncancer pain, reduced or absent CYP2D6 activity correlated with lack of therapeutic effect, whereas ultrarapid metabolizers showed an increased risk of AEs.47
Clinical practice guidelines published by the Clinical Pharmacogenetics Implementation Consortium may enable clinicians to use pharmacogenomic test results to optimize drug selection and dosing when a result is available (including opioids).48 However, pharmacogenomic testing for opioids remains controversial in cancer patients. Although CYP2D6 genotypes have clear effects on the pharmacokinetics of opioids such as oxycodone, they were not found to influence pain control, nausea, and sedation.49 Some researchers have postulated that the lack of correlation may involve several factors, including cachexia, which is associated with abnormalities in homeostasis that reflect a chronic inflammatory state in cancer patients, leading to differences in oxycodone metabolism.50
CONCLUSION
Pain is the most prevalent and debilitating symptom experienced by cancer patients. Initial and ongoing assessment of pain should be an integral part of supportive oncology. Opioids are the mainstay for management of moderate-to-severe cancer-related pain in the ambulatory-care setting. Adjuvants may be used as a single agent or added to opioids in selected patients. The use of pharmacogenomic testing for opioids remains controversial in cancer patients.
REFERENCES
1. International Association for the Study of Pain. IASP announces revised definition of pain. www.iasp-pain.org/PublicationsNews/NewsDetail.aspx?ItemNumber=10475. Accessed December 25, 2020.
2. Solano JP, Gomes B, Higginson IJ. A comparison of symptom prevalence in far advanced cancer, AIDS, heart disease, chronic obstructive pulmonary disease and renal disease. J Pain Symptom Manage. 2006;31(1):58-69.
3. Burton AW, Fanciullo GJ, Beasley RD, Fisch MJ. Chronic pain in the cancer survivor: a new frontier. Pain Med. 2007;8(2):189-198.
4. Hui D, Bruera E. A personalized approach to assessing and managing pain in patients with cancer. J Clin Oncol. 2014;32(16):1640-1646.
5. Fallon M, Giusti R, Aielli F, et al. Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2018;29(suppl 4):iv166-iv191.
6. Paice JA, Portenoy R, Lacchetti C, et al. Management of chronic pain in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2016;34(27):3325-3345.
7. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines). Adult cancer pain. Version 1.2020. www.nccn.org/professionals/physician_gls/pdf/pain.pdf. Accessed January 30, 2021.
8. Falk S, Dickenson AH. Pain and nociception: mechanisms of cancer-induced bone pain. J Clin Oncol. 2014;32(16):1647-1654.
9. Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69(1-2):1-18.
10. Hicks CL, von Baeyer CL, Spafford PA, et al. The Faces Pain Scale–Revised: toward a common metric in pediatric pain measurement. Pain. 2001;93(2):173-183.
11. Herr K, Spratt KF, Garand L, Li L. Evaluation of the Iowa Pain Thermometer and other selected pain intensity scales in younger and older adult cohorts using controlled clinical pain: a preliminary study. Pain Med. 2007;8(7):585-600.
12. Hølen JC, Hjermstad MJ, Loge JH, et al. Pain assessment tools: is the content appropriate for use in palliative care? J Pain Symptom Manage. 2006;32(6):567-580.
13. Manfredi PL, Breuer B, Meier DE, Libow L. Pain assessment in elderly patients with severe dementia. J Pain Symptom Manage. 2003;25(1):48-52.
14. Shang Y, Filizola M. Opioid receptors: structural and mechanistic insights into pharmacology and signaling. Eur J Pharmacol. 2015;763(pt B):206-213.
15. Bodnar RJ. Endogenous opiates and behavior: 2006. Peptides. 2007;28(12):2435-2513.
16. Kieffer BL, Evans CJ. Opioid receptors: from binding sites to visible molecules in vivo. Neuropharmacology. 2009;56(suppl 1):205-212.
17. Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol. 2018;25(1):4-12.
18. Gavériaux-Ruff C, Karchewski LA, Hever X, et al. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci. 2008;27(10):2558-2567.
19. Groer CE, Schmid CL, Jaeger AM, Bihn LM. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011;286(36):31731-31741.
20. Madariaga-Mazón A, Marmolejo-Valencia AF, Li Y, et al. Mu-opioid receptor biased ligands: a safer and painless discovery of analgesics? Drug Discov Today. 2017;22(11):1719-1729.
21. Pergolizzi J, Aloisi AM, Dahan A, et al. Current knowledge of buprenorphine and its unique pharmacological profile. Pain Pract. 2010;10(5):428-450.
22. Khanna IK, Pillarisetti S. Buprenorphine—an attractive opioid with underutilized potential in treatment of chronic pain. J Pain Res. 2015;8:859-870.
23. Siuda ER, Carr R III, Rominger DH, Violin JD. Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr Opin Pharmacol. 2017;32:77-84.
24. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain—United States, 2016. MMWR Recomm Rep. 2016;65(RR-1):1-49.
25. Cavallo J. Debating the role of opioids in the management of chronic cancer pain: a conversation with Leslie J Blackhall, MD, and Charles F von Gunten, MD, PhD. ASCO Post. https://ascopost.com/issues/may-25-2019/debating-the-role-of-opioids-in-the-management-of-chronic-cancer-pain/. Accessed December 22, 2020.
26. Boland JW, Bennett MI. State of the science: opioids and survival in cancer pain management. BMJ Support Palliat Care. 2020;10(4):379-380.
27. Anghelescu DL, Ehrentraut JH, Faughnan LG. Opioid misuse and abuse: risk assessment and management in patients with cancer pain. J Natl Compr Canc Netw. 2013;11(8):1023-1031.
28. Portenoy RK, Mehta Z, Ahmed E. Cancer pain management with opioids: optimizing analgesia. UpToDate. Waltham, MA: UpToDate; 2020. www.uptodate.com/contents/cancer-pain-management-with-opioids-optimizing-analgesia. Accessed December 23, 2020.
29. Caraceni A, Pigni A, Brunelli C. Is oral morphine still the first choice opioid for moderate to severe cancer pain? A systematic review within the European Palliative Care Research Collaborative guidelines project. Palliat Med. 2011;25(5):402-409.
30. Nicholson AB, Watson GR, Derry S, Wiffen PJ. Methadone for cancer pain. Cochrane Database Syst Rev. 2017;2(2):CD003971.
31. Anekar AA, Cascella M. WHO analgesic ladder. www.ncbi.nlm.nih.gov/books/NBK554435/. Accessed December 24, 2020.
32. Bennett MI. Effectiveness of antiepileptic or antidepressant drugs when added to opioids for cancer pain: systematic review. Palliat Med. 2011;25(5):553-559.
33. Filippiadis DK, Marcia S, Masala S, et al. Percutaneous vertebroplasty and kyphoplasty: current status, new developments and old controversies. Cardiovasc Intervent Radiol. 2017;40(12):1815-1823.
34. Portenoy RK, Copenhaver DJ. Cancer pain management: interventional therapies. UpToDate. Waltham, MA: UpToDate; 2020. www.uptodate.com/contents/cancer-pain-management-interventional-therapies#!. Accessed February 6, 2021.
35. Eidelman A, White T, Swarm RA. Interventional therapies for cancer pain management: important adjuvants to systemic analgesics. J Natl Compr Canc Netw. 2007;5(8):753-760.
36. Iacobucci G. NHS prescribed record number of antidepressants last year. BMJ. 2019;364:I1508.
37. Baldo BA. Opioid analgesic drugs and serotonin toxicity (syndrome): mechanisms, animal models, and links to clinical effects. Arch Toxicol. 2018;92(8):2457-2473.
38. Gillman PK. Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br J Anaesth. 2005;95(4):434-441.
39. Abadie D, Rousseau V, Logerot S, et al. Serotonin syndrome: analysis of cases registered in the French pharmacovigilance database. J Clin Psychopharmacol. 2015;35(4):382-388.
40. Jin Y, Desta Z, Stearns V, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97(1):30-39.
41. Haque R, Shi J, Schottinger JE, et al. Tamoxifen and antidepressant drug interaction in a cohort of 16,887 breast cancer survivors. J Natl Cancer Inst. 2015;108(3):djv337.
42. Azoulay L, Dell’Aniello S, Huiart L, et al. Concurrent use of tamoxifen with CYP2D6 inhibitors and the risk of breast cancer recurrence. Breast Cancer Res Treat. 2011;126(3):695-703.
43. Mercadante S, Bruera E. Opioid switching: a systematic and critical review. Cancer Treat Rev. 2006;32(4):304-315.
44. Mercadante S, Ferrera P, Villari P, et al. Frequency, indications, outcomes, and predictive factors of opioid switching in an acute palliative care unit. J Pain Symptom Manage. 2009;37(4):632-641.
45. Rennick A, Atkinson T, Cimino NM, et al. Variability in opioid equivalence calculations. Pain Med. 2016;17(5):892-898.
46. Samer CF, Daali Y, Wagner M, et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160(4):919-930.
47. Dagostino C, Allegri M, Napolioni V, et al. CYP2D6 genotype can help to predict effectiveness and safety during opioid treatment for chronic low back pain: results from a retrospective study in an Italian cohort. Pharmgenomics Pers Med. 2018;11:179-191.
48. Clinical Pharmacogenetics Implementation Consortium. Guidelines. https://cpicpgx.org/guidelines. Accessed December 25, 2020.
49. Andreassen TN, Eftedal I, Klepstad P, et al. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur J Clin Pharmacol. 2012;68(1):55-64.
50. Bugada D, Lorini LF, Fumagalli R, Allegri M. Genetics and opioids: towards more appropriate prescription in cancer pain. Cancers (Basel). 2020;12(7):1951.
Acknowledgment: The author would like to thank Deborah Chung for her assistance on this manuscript.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



