US Pharm. 2010;35(8):HS11-HS22.
Pulmonary hypertension (PH) is a serious and incurable condition characterized by high blood pressure in the arteries of the lungs. It was previously defined as mean pulmonary artery pressure (MPAP) >25 mmHg at rest or >30 mmHg with exercise, but the definition was simplified to resting MPAP ≥25 mmHg based on a review of current literature in 2008.1 Increased pulmonary artery pressure and resistance lead to right-sided heart failure and possible death if untreated. Life expectancy post PH diagnosis is less than 3 years for adults if left untreated.2 With 260,000 hospital visits and 15,668 deaths related to PH within the United States in 2002 alone, it is critical to identify and treat PH early.3,4

Clinical Classification of PH
The five-group World Health Organization (WHO) classification scheme was last updated in 2008 at the 4th World Symposium on Pulmonary Hypertension (TABLE 1).5 Patients in WHO Group 1 are classified as having pulmonary arterial hypertension (PAH), whereas patients in Groups 2 to 5 are classified as non-PAH or PH. Group 1 adds the criterion of pulmonary arterial wedge pressure of ≤15 mmHg and pulmonary vascular resistance (PVR) of ≥3 Wood's units.1 Group 1 also includes idiopathic and drug- and toxin-induced PAH. TABLE 2 lists the 2009 update of select agents and anorexigens associated with causing PAH.5 More substantial prevalence data are available for Group 1, but little is known for Groups 2 to 5. Prevalence is highly dependent on the underlying condition leading to PH. The prevalence of PAH is approximately 15 cases per million people with 6 cases per million people being idiopathic in nature.6 Idiopathic PAH (IPAH) occurs more commonly in women than in men (1.7:1). Although PAH can affect individuals of all ages, the mean age of presentation ranges from 36 to 50 years.2,6

Pathophysiology
Since PH in Groups 2 to 5 develops secondary to an underlying etiology, the discussion of pathophysiology in PH will focus on the mechanisms associated with PAH (Group 1) development, though they are not fully understood. Genetic mutations, polymorphisms, or alterations in biologic molecular pathways lead to endothelial dysfunction.7 Current drug therapies and research are centered on treating and correcting dysfunctional pathways, which include prostacyclin (PGI2), endothelin-1 (ET-1), and nitric oxide (NO) in vascular smooth muscle cells.8
In PAH, there is thought to be a decrease in prostacyclin synthase leading to imbalance of PGI2 expression compared to the vasoconstrictor thromboxane A2. This imbalance promotes increased platelet activation, thrombosis, proliferation, and vasoconstriction.7
ET-1 is a potent vasoconstrictor produced in the endothelium of pulmonary artery smooth muscle cells. ETA receptors on the endothelial cells stimulate vasoconstriction and proliferation while the ETB receptors are thought to stimulate the release of NO and PGI2, leading to vasodilation and counteracting the effects of ETA and ultimately ET-1. In PAH, less ET-1 is cleared. Higher levels of ET-1 correspond with more severe disease and prognosis.6
NO is a vasodilator that blocks calcium channels to exert effect. It is produced by endothelial smooth muscle cells via NO synthase. NO is necessary for the conversion of cyclic guanosine triphosphate to cyclic guanosine monophosphate (cGMP) by guanylate cyclase to promote smooth muscle cell relaxation. In PAH, the enzyme phosphodiesterase-5 (PDE-5), found largely in the lung, inactivates cGMP, preventing cellular response, causing pulmonary smooth muscle vasoconstriction, and leading to platelet activation and proliferation. Vasoconstriction also predominates due to dysfunctional calcium channel inhibition and less NO synthase.6,7
Other potential mechanisms for PAH include autoantibodies and inflammatory processes. Alterations in norepinephrine and serotonin systems are believed to play a role in the development of anorexigenic PAH; however, this alone is not thought to cause PAH as its incidence has not risen with increasing use of selective serotonin reuptake inhibitors (SSRIs).7
Presentation
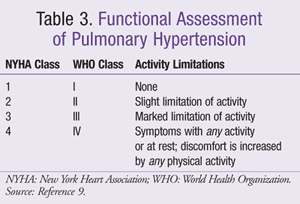
Signs and symptoms of PH vary based on comorbidities and disease severity.7 Dyspnea is the most frequently reported symptom. Disease severity may be determined by first assessing exercise capacity or functionality as defined by WHO Functional Assessment Classification and/or New York Heart Association (NYHA) Functional Classification (TABLE 3).9 Exercise capacity, often determined by the 6-minute walk (6MW) test, serves as marker for disease severity, progression, and treatment response. Second, echocardiography assesses disease progression, but right heart catheterization is considered the gold standard since it is a more accurate measure of hemodynamic parameters and vasoreactivity. This test is more invasive and is thus reserved for advanced treatment.7,10 Goals of therapy are to reduce or alleviate symptoms, improve quality of life, improve functional class, and increase survival.6,12,13
Treatment Options
Nonpharmacologic management includes measures to improve or prevent worsening functional status. This includes restricting fluid intake to <1.5 liters daily and sodium intake to <2,400 mg daily and participating in cardiopulmonary rehabilitation.6,7,10,11
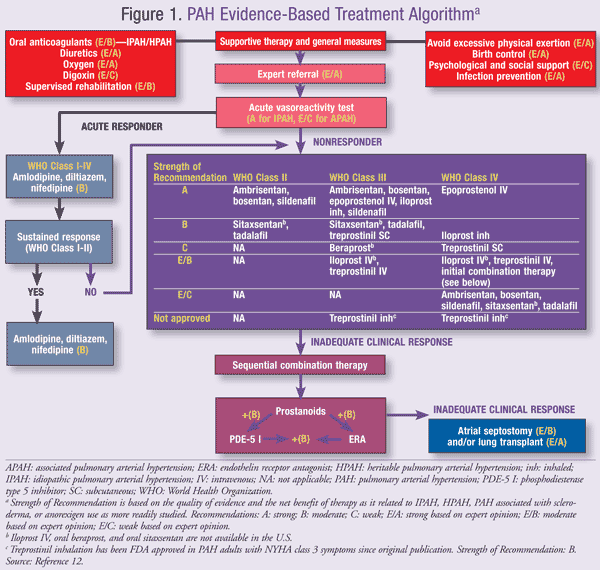
Pharmacologic treatment encompasses primary and advanced therapy (FIGURE 1).12 Primary therapy for PH, also referred to as standard or supportive therapy, is guided by identifying and treating the underlying cause. It may be utilized in all PH groups, providing symptomatic relief for WHO functional class II, III, and IV (TABLE 3).9 Advanced therapy focuses on preventing disease progression by treating underlying cardiopulmonary disorders. Patients refractory to treatment may require lung and/or heart transplantation.
Supportive therapy includes oral anticoagulants, diuretics, oxygen, and digoxin. Anticoagulation with warfarin may be useful in patients with or at risk for venous thromboembolism PH (Group 4) and for PAH (Group 1) patients with idiopathic, hereditary, and drug-induced or anorexigen-induced PAH, portal hypertension, scleroderma, or congenital heart disease. The target international normalized ratio range is 1.5 to 3.0 depending on the indication.6-10 Evidence for anticoagulant use stems from observational studies where survival benefit was noted.6 For fluid overload and hepatic congestion, loop diuretics are recommended and adjusted to response. Supplemental oxygen of 1 to 4 L/min is utilized to maintain oxygen saturations above 90% at all times and prevent vasoconstriction due to hypoxemia. Oxygen is widely used in PH due to pulmonary diseases and/or hypoxia (Group 3). Lastly, the role of digoxin is not clearly defined in Group 1 PAH. Digoxin may be useful in Group 3 patients with chronic obstructive pulmonary disease (COPD) and biventricular heart failure.6-10
Advanced therapy is not indicated or well studied in PH Groups 2 to 5; therefore, these patients should receive treatment for underlying cardiac, pulmonary, or vascular conditions when possible. Group 1 PAH is primarily idiopathic in nature and therefore lacks an underlying cause.13 The subsequent sections will focus on Group 1 PAH treatment in adults where advanced therapy has been developed to prevent or slow progression when used with primary therapy.
Pharmacologic Management of PAH
Calcium Channel Blockers (CCBs): Advanced therapy is based on severity of disease as determined by right heart catheterization and responsiveness to vasoactivity testing with epoprostenol (2-10 ng/kg/min), adenosine (50-250 mcg/kg/min), or NO (10-80 parts per million [ppm] for 5 minutes).6 Currently there are no evidence-based guidelines for selecting a vasodilatory testing agent. Positive response to vasoreactivity testing is a reduction in MPAP by ³10 mmHg to achieve an MPAP of ≤40 mmHg, which may indicate responsiveness to CCBs.6 It may also indicate earlier stages of disease and thus a better prognosis or other underlying condition treatable with CCBs such as IPAH or anorexigen-induced PAH. In an early study, several patients deemed responders to vasoactive testing improved from NYHA functional class 3 to functional class 1 or 2.14 A subsequent study showed CCB therapy in responders extended predicted survival over 5 years (predicted survival 40% without therapy versus 90% with CCB therapy).
Although not FDA approved for the treatment of PAH, CCBs commonly used are long-acting nifedipine, sustained-release diltiazem (useful for tachycardia), and amlodipine. Once-daily doses of nifedipine 120 to 240 mg, diltiazem 540 to 900 mg, and amlodipine 2.5 mg to 40 mg have been successfully used. All agents are titrated every 2 to 4 weeks to clinical effect. Verapamil should be avoided due to negative inotropic effects.14,15
Prostacyclin Analogs: If a patient does not sustain a response to CCBs after at least 3 to 6 months of therapy or is deemed a nonresponder to vasoactivity testing, drug therapy with prostacyclins, endothelin receptor antagonists, and/or PDE-5 inhibitors is initiated.6,12 (A summary of FDA-approved treatments for PAH is provided in TABLE 4).16-23 Three prostacyclin analogs are currently available in the U.S.—epoprostenol (Flolan), treprostinil (Remodulin and Tyvaso), and iloprost (Ventavis).
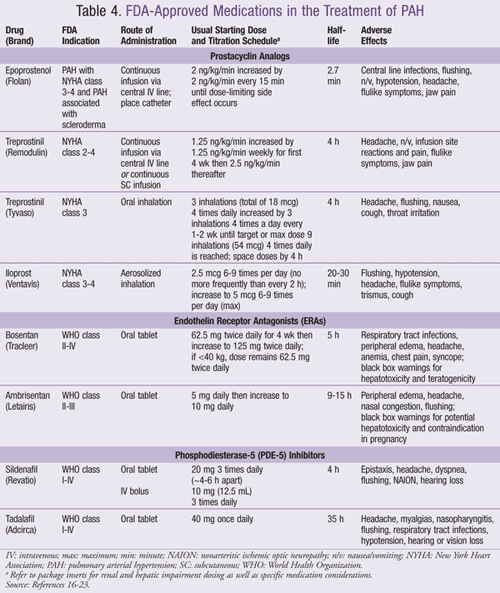
Intravenous (IV) epoprostenol is the only prostacyclin to improve survival in WHO class III and IV patients in studies compared to supportive therapy alone.24-26 Patients receiving epoprostenol plus standard therapy had 1-, 3-, and 5-year survival rates of 87%, 63%, and 54%, respectively, compared to the historical control group of 77%, 41%, and 27% (P = .045).24,25 Similar results for survival were found in an observational study in NYHA class 3 and 4 PAH patients treated with epoprostenol and primary therapy.26 Epoprostenol has also been shown to improve functional capacity, quality of life, hemodynamic parameters, and survival with primary therapy compared to primary therapy alone.24-26 Epoprostenol is considered first-line therapy for WHO class III and IV. It is also the gold standard for end-stage PH.10 Epoprostenol, like other FDA-approved medications for PAH, must be obtained through a specialty pharmacy distribution program. Due to its short half-life, interrupting drug delivery may lead to rebound PH or death.16
Treprostinil, the second FDA-approved prostacyclin, offers IV, subcutaneous (SC), and inhaled formulations. Severe infusion site pain and paresthesias appeared to limit dose titration in studies and led to therapy discontinuation of the SC formulation.7 The IV formulation is useful in patients who do not tolerate the SC infusion. The IV and SC formulations are bioequivalent and are used in treating patients with NYHA class 2, 3, and 4 PAH.10 The inhalation treprostinil (Tyvaso) received FDA approval for PAH patients with NYHA class 3 symptoms only. Controlled studies demonstrated a mean 20-meter improvement in 6MW distance at 12 weeks when added to bosentan or sildenafil therapy.18 This formulation may be suited for an ambulatory patient intolerant of other PAH medications or in whom symptoms persist despite current therapy. All three formulations have shown an approximate 20-meter improvement in 6MW test. The SC and IV formulations are better studied and have demonstrated improvements in hemodynamic parameters and symptoms with dose increases.27
Gram-negative bacterial infections have been noted with IV treprostinil, as well as epoprostenol, due to the IV delivery method. Other side effects of treprostinil are similar to that of epoprostenol; however, the safety profile of treprostinil is more attractive than epoprostenol. Rebound PH is less likely to occur with interruptions in therapy or dysfunction of treprostinil delivery devices since it has a longer half-life than epoprostenol.16-18
Iloprost is an aerosolized prostacyclin analog approved for treating PAH Group 1 with NYHA functional class 3 or 4 symptoms.19,28 It is dosed 6 to 9 times per day. Iloprost efficacy appears to be limited and wanes over time. Based on the lack of strong efficacy data, iloprost may be reserved for patients who are unable or unwilling to take IV or SC PAH agents or in whom oral agents are not effective.6,8,9
Endothelin Receptor Antagonists (ERAs): Besides the prostacyclin pathway, the FDA has also approved the ERAs bosentan (Tracleer) and ambrisentan (Letairis) to promote vasodilation of pulmonary vascular smooth muscle cells (TABLE 4). Bosentan is an oral nonselective ERA indicated for PAH with WHO class II, III, and IV symptoms to improve exercise capacity and decrease clinical worsening of disease based on randomized, double-blind, placebo-controlled trials.29 A randomized, controlled bosentan trial was the first to define and demonstrate longer time to clinical worsening, defined as time to death, lung transplantation, PH hospitalization, lack of clinical improvement, or worsening leading to discontinuation of therapy, need for epoprostenol therapy, or atrial septostomy. (The definition may vary slightly in other studies.) Studies also showed improvement in functional class, cardiac index, PVR, and MPAP.29
Once-daily ambrisentan is an ETA receptor selective oral ERA. Receptor selectivity is thought to be more favorable promoting vasodilation. It is indicated for PAH patients with WHO class II and III symptoms to prolong time to clinical worsening and improve exercise capacity and hemodynamic parameters.30 Potential for interactions exist as ambrisentan and bosentan are metabolized by CYP2C9 and 3A4 pathways. Hematocrit must be monitored with both agents. Due to hepatotoxicity and teratogenicity, patients must enroll in a monitoring program with bosentan (Tracleer Access Program [TAP]), and ambrisentan has a special distribution protocol (Letairis Education and Access Program [LEAP]). Monthly pregnancy tests should also be obtained.20,21
PDE-5 Inhibitors: Sildenafil (Revatio) and tadalafil (Adcirca) demonstrate improved exercise capacity in patients with PAH. The published sildenafil and tadalafil studies were not designed to show mortality benefit. Sildenafil, oral or IV, is FDA approved to improve exercise capacity and delay clinical worsening in patients with PAH.22,31 Patients treated with sildenafil had statistically significant increases in their 6MW test compared to those treated with placebo (>38 m). Additionally, as compared to placebo, more patients treated with sildenafil improved by at least one NYHA functional class after the 12-week treatment period. Most patients were NYHA functional class 2 or 3 at baseline. Effects were unchanged after 12 months of treatment. The changes in the 6MW distance with sildenafil are comparable to changes observed in patients treated with IV epoprostenol (47 m), inhaled iloprost (36 m), and oral bosentan (44 m), although these agents have not been directly compared.32
Tadalafil is FDA approved to improve exercise capacity in patients with WHO Group 1 PAH.23 Galiè et al found patients randomized to tadalafil 40 mg once daily for 16 weeks had a mean increase in 6MW distance by 44 more meters than placebo (P <.01).33 Adverse effects with tadalafil were similar to sildenafil (TABLE 4).
Combination Therapy: Combination therapy is being studied for targeting the different pathologic processes of PAH. Data both support and refute statistically significant improvement in functional capacity. Several studies failed to demonstrate statistical significance due to lack of power owing to small sample size or smaller-than-expected improvement in combination therapy.34-36 Combinations studied include prostacyclin plus an ERA, prostacyclin plus a PDE-5 inhibitor, and an ERA plus a PDE-5 inhibitor. Inhaled iloprost, when added to background bosentan, increased 6MW test results by approximately 30 meters compared to a 4-meter increase in placebo plus bosentan (P = .051). After 12 weeks of treatment, functional class improvement was seen in 34% of iloprost plus bosentan treated patients with baseline NYHA class 2 or 3 (P = .002). In addition, time to clinical worsening was delayed in the iloprost treatment group.34 The 6MW distance increased 29.8 meters in IPAH or associated PAH patients on epoprostenol plus sildenafil versus epoprostenol plus placebo (P <.001). Improvements in hemodynamics and a longer time to clinical worsening in the combination group were also seen.34 When initiating combination therapy, providers should consider side-effect profiles and dose-limiting side effects. In theory, lower doses of each agent may be used to limit dose-related side effects; however, side effects were reported more frequently with combination therapy (epoprostenol plus bosentan, iloprost plus bosentan) except for epoprostenol plus sildenafil versus epoprostenol alone.34-37
Conclusion
While there is no cure for PAH (WHO Group 1), prostacyclins, ERAs, PDE-5 inhibitors, and supportive care have been shown to reduce symptoms of this condition. Despite interventions, over time patients will experience worsening of functional class, symptoms, quality of life, and hemodynamic parameters. Therapy selection should be based on evidence, NYHA functional class, ease of administration, tolerability of side-effect profile, drug interaction potential, and cost. For patients with more functionality, therapy with oral agents may be more reasonable to try first. As functionality worsens, prostacyclins (i.e., epoprostenol) may be more appropriate to utilize as there is greater ability to titrate to desired effect. Epoprostenol has also shown survival benefit in studies. Additional counseling should be provided to patients regarding device safety, drug therapy monitoring, pregnancy testing, and immunizations (pneumococcal, H1N1, and influenza). Patients should receive follow-up care from a center specialized in treating patients with PAH and be seen at least every 3 to 6 months for reevaluation of therapy.6
As for PH (Groups 2 to 5), treatment of the underlying cause with supportive care slows disease or treats disease progression. The use of prostacyclins, ERAs, PDE-5 inhibitors, or a combination of these products is not indicated for use in Groups 2 to 5, as there is a lack of supportive evidence in the current studies. Future studies are needed to better define PAH and PH and prevent or prolong time to clinical worsening.
REFERENCES
1. Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S55-S66.
2. Machado RD, Eickelberg O, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S32-S42.
3. Hyduk A, Croft JB, Ayala C, et al. Pulmonary hypertension surveillance—United States, 1980-2002. MMWR. 2005;54:1-28.
4. Barst RJ. Pulmonary hypertension: Past, present and future. Ann Thorac Med. 2008;3:1-4.
5. Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S43-S54.
6. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119:2250-2294.
7. Talbert RL, Boudreaux R, Owens RL. Pulmonary hypertension. In: Dipiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 7th ed. New York, NY: McGraw-Hill; 2008:519-520.
8. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425-1436.
9. Rubin LJ; American College of Chest Physicians. Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(suppl 1):7S-10S.
10. Rubin LJ, Hopkins W. Overview of pulmonary hypertension. In: UpToDate. Mandel J, ed. Waltham, MA: UpToDate; 2009.
11. Varney KL. Pulmonary arterial hypertension. In: Richardson M, Chant C, Cheng JW, et al, eds. Pharmacotherapy Self-Assessment Program. 6th ed. Lenexa, KS: American College of Clinical Pharmacy; 2008:163-182.
12. Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S78-S84.
13. Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S55-S66.
14. Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76:135-141.
15. Rich S, Kaufmann E, Levy P. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76-81.
16. Flolan (epoprostenol) package insert. Research Triangle Park, NC: GlaxoSmithKline; January 2008.
17. Remodulin (treprostinil) package insert. Research Triangle Park, NC: United Therapeutics Corp; January 2010.
18. Tyvaso (treprostinil) package insert. Research Triangle Park, NC: United Therapeutics Corp; July 2009.
19. Ventavis (iloprost) package insert. South San Francisco, CA: Actelion Pharmaceuticals US, Inc; April 2010.
20. Tracleer (bosentan) package insert. South San Francisco, CA: Actelion Pharmaceuticals US Inc; August 2009.
21. Letairis (ambrisentan) package insert. Foster City, CA: Gilead Sciences, Inc; August 2009.
22. Revatio (sildenafil) package insert. New York, NY: Pfizer Labs; November 2009.
23. Adcirca (tadalafil) package insert. Indianapolis, IN: Eli Lilly and Company; May 2009.
24. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296-301.
25. Sandoval J, Bauerle O, Palomar A, et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation. 1994;89:1733-1744.
26. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477-1482.
27. Simonneau G, Barst RJ, Galiè N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind randomized controlled trial. Am J Respir Crit Care Med. 2002;165:800-804.
28. Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322-329.
29. Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119-1123.
30. Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010-3019.
31. Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148-2157.
32. Barst RJ, Rubin LJ, McGoon MD, et al. Survival in primary pulmonary hypertension with long-term continuous infusion prostacyclin. Ann Intern Med. 1994;121:409-415.
33. Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894-2903.
34. McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:1257-1263.
35. Simonneau G, Rubin LJ, Galiè N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149:521-530.
36. Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24:353-359.
37. Hoeper MM, Leuchte H, Halank M, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2006;28:691-694.
To comment on this article, contact rdavidson@uspharmacist.com.



