US Pharm. 2007;32(3):HS-5-HS-32.
New
molecular entities (NMEs), as defined by the FDA, are new drug products that
have as their active ingredient a chemical substance marketed for the first
time in the United States. The following descriptions of the NMEs approved
during the second half of 2006 detail the pharmacotherapeutic design and
mechanism of action of each new drug. Also included is a summary of selected
clinical data presented to the FDA in support of the manufacturer's new drug
application (NDA). The FDA classifies NMEs on the basis of therapeutic
potential (Table). NMEs classified as priority reviewed (P) represent
significant improvement compared to marketed products in the treatment,
diagnosis, or prevention of a disease. NMEs receiving standard review (S) are
those that appear to have therapeutic qualities similar to those of one or
more already marketed drugs.
This review is intended to be
objective rather than evaluative in content. The information for each reviewed
NME was obtained primarily from sources published prior to FDA approval.
Experience clearly demonstrates that many aspects of a new drug's therapeutic
profile, not detected in premarketing studies, surface after the drug is used
in large numbers of patients. Studies have clearly demonstrated the appearance
of "new" adverse reactions for many NMEs within two to three years of the drug
becoming available. Many of these drugs may eventually acquire at least one
black box warning for serious adverse drug reactions or are withdrawn from the
market for safety reasons that were not recognized at the time of approval.
Hence, while this review offers a starting point for learning about new drugs,
it is essential that practitioners be vigilant of changes in a drug's
therapeutic profile as reported by their own patients and in the
pharmaceutical literature.

Posaconazole (Noxafil, Schering)
Introduction:1-4
Oropharyngeal candidiasis (OPC) is an opportunistic mucosal infection caused
by Candida species, in most cases Candida albicans. The four
major forms of OPC are pseudomembranous, or thrush, consisting of white
discrete plaques on an erythematous background, on the buccal mucosa, throat,
tongue, or gingivae; erythematous, consisting of smooth red patches on
the hard or soft palate, dorsum of the tongue, or buccal mucosa; hyperplastic,
consisting of white, firmly adherent patches or plaques, usually bilaterally
distributed on the buccal mucosa; and denture-induced stomatitis, presenting
as either a smooth or granular erythema confined to the denture-bearing area
of the hard palate and often associated with angular cheilitis. Symptoms vary,
ranging from no symptoms to a sore and painful mouth with a burning tongue and
altered taste. OPC can also impair speech, nutritional intake, and quality of
life.
Candida is found in the
mouths of 31% to 60% of healthy people in developed countries, and denture
stomatitis associated with Candida is present in 65% of denture
wearers. OPC can affect up to 15% to 60% of people with hematologic or
oncological malignancies during periods of immunosuppression. This condition
is also seen in 7% to 48% of people with HIV infection and in over 90% of
those with AIDS. Risk factors associated with symptomatic OPC, in addition to
immunosuppression and hematologic disorders, include broad-spectrum antibiotic
use, inhaled or systemic steroid use, xero stomia, diabetes, and the use
of dentures, obturators, or orthodontic appliances. For most people, untreated
candidiasis persists for months or years unless risk factors are treated or
eliminated.
A number of therapeutic
options are available for the treatment of OPC. Topical therapy with
antifungal preparations, including nystatin lozenges or suspension or
clotrimazole troches, usually suffices for mild forms of the disease. However,
extensive disease, especially in patients with immunosuppression (from cancer
or HIV/AIDS), and disease in which there are symptoms that suggest esophageal
involvement (e.g., pain on swallowing) are best treated with systemic
antifungal therapy; prolonged suppressive therapy may be required if the
immunosuppressive condition does not remit. Historically, the primary systemic
therapies applied for OPC employ an azole antifungal such as fluconazole,
itraconazole, or ketoconazole. Although OPC is usually amenable to therapy
with local or systemic antifungal drugs, failures of azole therapy for such
infections have been reported, and relapse rates are high (30% to 50%). For
example, fluconazole-refractory candidiasis reportedly occurs in 5% to10% of
HIV-infected patients with low CD4+ counts who have received chronic treatment
with fluconazole. Strains isolated during relapses are probably mutants of
previously present susceptible strains of C albicans. Thus, there
remains a need for new therapies to effectively treat OPC, especially in
patients who are immunocompromised or have a hematologic disorder.
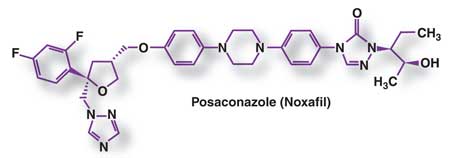
In September 2006, the FDA
approved posaconazole (Figure) as an oral suspension for the treatment
of OPC, including infections refractory to itraconazole and/or fluconazole.
This action followed an approval earlier in the year for prevention of
invasive Aspergillus and Candida infections in patients ages 13
and older who are at high risk of these infections due to being severely
immunocompromised, such as hematopoietic stem cell transplant (HSCT)
recipients with graft-versus-host disease (GVHD) or those with hematologic
malignancies with prolonged neutropenia from chemotherapy. Invasive fungal
infections are a leading cause of death in these high-risk populations.
Mechanism of Action:2-5
Like other azole antifungals, posaconazole blocks the synthesis of
ergosterol, a key component of the fungal cell membrane, through inhibition of
the enzyme lanosterol 14-alpha-demethylase and the accumulation of methylated
sterol precursors.
Posaconazole has demonstrated
in vitroactivity against Aspergillus fumigatus and C albicans.
However, correlation between the results of susceptibility studies and
clinical outcomes has not been established, and interpretive
criteria/breakpoints for posaconazole have not been established for any fungi.
In immunocompetent and/or immunocompromised mice and rabbits with pulmonary or
disseminated infection with A fumigatus, prophylactic administration of
posaconazole was effective in prolonging survival and reducing mycological
burden. Prophylactic posaconazole also prolonged survival of immunocompetent
mice challenged with C albicans or Aspergillus flavus. Clinical
isolates of C albicans and Candida glabrata with decreased
posaconazole susceptibility were observed in oral samples taken during
prophylaxis with posaconazole and fluconazole, suggesting a potential for
resistance. These isolates also showed reduced susceptibility to other azoles,
indicating possible cross-resistance between azoles.
Pharmacokinetics:3,5
Posaconazole is absorbed with a median time to maximum plasma concentration
(Tmax) of approximately three to five hours. Dose-proportional increases in
plasma exposure (area under the curve [AUC]) to posaconazole were observed
following single oral doses from 50 to 800 mg and following multiple-dose
administration from 50 mg two times a day to 400 mg two times a day. No
further increases in exposure were observed with dosages greater than 400 mg
twice daily. In single-dose studies, the mean AUC and maximum plasma levels
(Cmax) of posaconazole were approximately three times higher when administered
with a nonfat meal or liquid nutritional supplement (14 g of fat) and about
four times higher when administered with a high-fat meal (~50 g fat) relative
to the fasted state. Thus, to ensure attainment of adequate plasma
concentrations, it is recommended to administer posaconazole with food or a
nutritional supplement. Posaconazole is highly protein-bound (>98%),
predominantly to albumin, and has an apparent volume of distribution of 1,774
L, suggesting extensive extravascular distribution and penetration into the
body tissues.
Posaconazole does not undergo
any significant degree of oxidative (cytochrome P450 [CYP450]-mediated)
metabolism but is conjugated to some degree by uridine diphosphate (UDP)
glucuronidation (phase 2 enzymes). Posaconazole is eliminated primarily in the
feces (71%), with the major component eliminated as parent drug (66% of dose).
Renal clearance is a minor elimination pathway, with 13% of the dose excreted
in urine in up to 120 hours. Excreted metabolites in both urine and feces
account for about 17% of the administered posaconazole dose. The mean
elimination half-life is 35 hours (range, 20 to 66 hours) with a total body
clearance (CL/F) of 32 L/hour.
The pharmacokinetics of
posaconazole are not altered significantly in mild (creatinine clearance
[ClCr] 50 to 80 mL/min/1.73 m2) and moderate (ClCr 20 to 49
mL/min/1.73 m2) renal impairment. Thus, no dose adjustment is
required in these patients. In patients with severe renal insufficiency (ClCr
<20 mL/min/1.73 m2), the mean AUC was similar to that in
patients with normal renal function, but the range of AUC estimates was highly
variable. Due to variability in exposure, patients with severe renal
impairment should be monitored closely for breakthrough fungal infections.
Currently, there is inadequate pharmacokinetic data in patients with hepatic
impairment to be able to determine if dose adjustment is necessary. Thus, it
is recommended that posaconazole be used with caution in patients with hepatic
impairment. The pharmacokinetic profile of posaconazole is not affected
significantly by gender, race, or age.
Clinical Profile:3,6-8
Posaconazole is indicated for prophylaxis of invasive Aspergillus and
Candida infections in patients ages 13 and older who are at high risk due
to a severely immunocompromised state. The efficacy of posaconazole in the
prophylaxis of invasive fungal infections was demonstrated in two randomized,
controlled studies (designated as study 1 and study 2) in more than 1,000
patients with severely immunocompromised immune systems. In both trials,
aspergillosis was the most commonly observed breakthrough infection.
In study 1, patients with
neutropenia receiving cytotoxic chemotherapy (n = 602) were randomized to
receive posaconazole three times daily or pooled standard azole therapy
(fluconazole oral suspension 400 mg once daily or itraconazole oral solution
200 mg twice daily). Results showed that posaconazole prophylactic therapy
yielded a reduction in treatment failure, compared to
fluconazole/itraconazole, as defined by a composite end point of breakthrough
invasive fungal infections, death, and use of systemic antifungal drugs (27%
vs. 42%). Posaconazole was associated with lower rates of proven or probable
invasive fungal infections (2% vs. 8%), a reduced number of breakthrough
Aspergillus infections (1% vs. 7%), and decreased all-cause mortality
rates at 100 days postrandomization (14% vs. 21%).
Study 2 compared posaconazole
suspension (200 mg three times daily) with fluconazole capsules (400 mg once
daily) for invasive fungal-infection prophylaxis in allogenic HSCT recipients
with GVHD (n = 600). In this trial, posaconazole was shown to significantly
reduce the rate of proven or probable invasive fungal infections and related
mortality at 16 weeks (5% vs. 9% and 3% vs. 5%, respectively).
More recently, the FDA has
approved posaconazole for the treatment of OPC, including OPC refractory to
itraconazole and/or fluconazole, based on results primarily from two
additional trials. One trial, designated as study 3, was a randomized,
controlled, evaluator-blinded study in HIV-infected patients with OPC in which
the majority of subjects were infected with C albicans. Patients were
treated with posaconazole or fluconazole oral suspension, both drugs
administered as 100 mg twice daily for one day, followed by 100 mg once daily
for 13 days). Clinical and mycological outcomes were assessed after 14 days of
treatment and at four weeks after the end of treatment. Patients who received
at least one dose of study medication and had a positive oral swish culture of
Candida species at baseline were included in the analyses. Posaconazole
therapy achieved similar rates of clinical success (complete or partial
resolution of all ulcers and/or plaques and symptoms) and mycological
eradication (absence of colony-forming units) at 14 days, compared to
fluconazole (91.7% vs. 92.5% and 52.1% vs. 50.0%, respectively). Clinical and
mycological relapse rates at four weeks posttherapy were also comparable
between groups (29.0% vs. 35.1% and 55.6% vs. 63.7%, respectively).
A second study (designated as
study 4) was a noncomparative study of posaconazole oral suspension in
HIV-infected subjects with OPC that was refractory to treatment with
fluconazole or itraconazole. An episode of OPC was considered refractory if
there was a lack of improvement or a worsening of OPC after a standard course
of therapy with fluconazole at doses ?100 mg/day for at least 10
consecutive days or itraconazole 200 mg/day for at least 10 consecutive days,
and treatment with fluconazole or itraconazole had not been discontinued for
more than 14 days prior to treatment with posaconazole. Of the 199 subjects
enrolled in this study, 89 subjects met these criteria for refractory
infection. Forty-five subjects with refractory OPC were treated with
posaconazole 400 mg twice daily for three days, followed by 400 mg once a day
for 25 days, with an option for further treatment during a three-month
maintenance period. Following a dosing amendment, 44 more subjects were
treated with posaconazole 400 mg twice daily for 28 days. The efficacy of
posaconazole was assessed by the clinical success (cure or improvement) rate
after four weeks of treatment. The clinical success rate was 74.2% (66/89).
The clinical success rates for both the original and amended dosing regimens
were similar (73.3% and 75.0%, respectively).
Adverse Reactions:3,6-8
To date, the safety of posaconazole has been assessed in more than 2,000
patients in clinical trials. The most commonly reported adverse events
associated with posaconazole are fever (45%), gastrointestinal (GI) effects
(27% to 42%; e.g., diarrhea, nausea, vomiting, abdominal pain, constipation),
hypokalemia (30%), headache (28%), coughing (24%), rigors (20%), dyspnea
(20%), rash (19%), hypertension (18%), fatigue (17%), and blood disorders (20%
to 29%; e.g., anemia, neutropenia, thrombocytopenia). Rare cases of hepatic
reactions (e.g., mild-to-moderate elevations in alanine aminotransferase
[ALT], aspartate aminotransferase [AST], alkaline phosphatase, total
bilirubin, and/or clinical hepatitis) have also been reported in association
with posaconazole. Thus, liver function tests should be evaluated at the
beginning and during the course of posaconazole therapy.
Serious, or medically
significant, rare treatment-related adverse events reported during clinical
trials with posaconazole have been adrenal insufficiency, allergic, and/or
hypersensitivity reactions. In addition, rare cases of hemolytic uremic
syndrome, thrombotic thrombocytopenic purpura, and pulmonary embolus have been
seen primarily among patients who had received concomitant cyclosporine or
tacrolimus for the management of transplant rejection or GVHD. During clinical
development, there was a single case of torsade de pointes in a patient taking
posaconazole. This report involved a seriously ill patient with multiple
confounding, potentially contributory risk factors, such as a history of
palpitations, recent cardiotoxic chemotherapy, hypokalemia, and
hypomagnesemia.
Drug Interactions:3,6-8
A number of azole antifungals, such as ketoconazole, depend on an acidic
environment for optimal dissolution and absorption from the GI tract.
Therefore, drugs that decrease gastric acid secretion (e.g., antacids, H2
-antagonists, proton pump inhibitors) can impair absorption of some azoles. No
clinically significant effects on bioavailability or plasma concentrations
were observed when posaconazole was administered with an antacid, proton pump
inhibitor, or H2-antagonist other than cimetidine. Since cimetidine
significantly reduces posaconazole absorption, its concurrent use should be
avoided.
Posaconazole is primarily
metabolized via UDP glucuronidation (phase 2 metabolism) and is a substrate
for p-glycoprotein efflux pump. Therefore, inhibitors or inducers
(rifabutin, phenytoin) of these clearance pathways may affect posaconazole
plasma concentrations. Clinical studies and in vitro studies with human
hepatic microsomes indicate that posaconazole is an inhibitor of cytochrome
enzymes, particularly the CYP3A4 isozyme. Therefore, posaconazole has the
potential to increase plasma concentrations of other drugs that are
predominantly inactivated by CYP3A4 metabolism. This can be particularly
significant when the other drug is a CYP3A4 substrate with a narrow
therapeutic index, such as cyclosporine, tacrolimus, midazolam, rifabutin, and
phenytoin. Based on this potential interaction, drug dosages may need to be
reduced at the initiation of posaconazole treatment, and blood concentrations
may need to be monitored frequently during, and at discontinuation of,
posaconazole administration. The manufacturer's literature should be consulted
for specific dosing and monitoring recommendations when initiating
posaconazole therapy in patients receiving other drugs.
Dosage and Administration:
3 Posaconazole is supplied as an oral suspension in 4-oz. (123-mL)
amber glass bottles containing 105 mL of suspension (40 mg of posaconazole per
milliliter). The product should be shaken well before use and administered as
400 mg (10 mL) twice a day for OPC. Duration of therapy should be based on the
severity of the patient's underlying disease and clinical response. A measured
dosing spoon is provided, marked for doses of 2.5 and 5 mL. To enhance oral
absorption and optimize plasma concentrations, the drug should be administered
with a full meal or with a liquid nutritional supplement in patients who
cannot eat a full meal. Patients who experience severe diarrhea or vomiting
should be monitored closely for breakthrough fungal infections. In addition,
coadministration of drugs that can decrease the plasma concentrations of
posaconazole should be generally avoided unless the benefit outweighs
the risk. If such drugs are necessary, patients should be monitored closely
for breakthrough fungal infections. The duration of therapy is based on
recovery from neutropenia or immunosuppression.
No dosage adjustment is
recommended for patients with renal dysfunction. However, due to the high
variability in exposure, patients with severe renal impairment should be
monitored closely for breakthrough fungal infections. The pharmacokinetic data
in subjects with hepatic impairment were not sufficient to determine if dosage
adjustment is necessary in patients with hepatic dysfunction. It is
recommended that posaconazole be used with caution in patients with hepatic
impairment.
Dasatinib (Sprycel, Bristol-Myers Squibb)
Introduction and Mechanism of
Action:9-11 Chronic myelogenous leukemia (CML) is a cancer of
blood cells characterized by replacement of the bone marrow with malignant,
leukemic cells called blast cells. As the number of blast cells
increases in the blood and bone marrow, there is a decrease in white blood
cells, red blood cells, and platelets. This may result in infections, anemia,
and bleeding, as well as in bone pain and pain or a feeling of fullness below
the ribs on the left side. The number of blast cells in the blood and bone
marrow and the severity of symptoms determine the phase of CML. The three
phases of CML are chronic, accelerated, and blastic. In chronic-phase CML,
fewer than 10% of the cells in the blood and bone marrow are blast cells; in
the accelerated phase, 10% to 19% are blast cells. In blastic-phase CML, 20%
or more of the cells in the blood or bone marrow are blast cells. Blastic
transformation may be myeloid, lymphoid, undifferentiated, or mixed,
with myeloid blast crisis being about two times more common than
lymphoid. A blast crisis consists of fatigue, fever, and an enlarged spleen
that occur during the blastic phase. Diagnosis of the CML phase is important
to determine therapy.
CML is usually diagnosed by
finding a specific chromosomal abnormality, the Philadelphia (Ph) chromosome,
named after the city where it was first recorded. The Ph chromosome is the
result of the exchange of genetic material between the long arms of
chromosomes 9 and 22, a process referred to as translocation. This
exchange brings together two genes: the BCR (breakpoint cluster region) gene
on chromosome 22 and the proto-oncogene ABL (Abelson leukemia virus) on
chromosome 9. The resulting hybrid gene BCR-ABL codes for a fusion protein
with tyrosine kinase activity, which activates signal transduction pathways,
leading to uncontrolled cell growth.
Imatinib, an orally available
ABL kinase inhibitor, can induce hematologic and cytogenetic
remission in all stages of CML, as well as in Ph-positive acute
lymphoblastic leukemia (Ph+ALL), with minimal toxicity. Imatinib is
now first-line therapy for newly diagnosed CML. However, resistance
to imatinib has become increasingly important. Furthermore, nearly all
patients with chronic-phase CML have persistent disease, measurable
by polymerase chain reaction and indicative of a reservoir of
residual leukemia cells that may be a source of relapse. Relapse
during imatinib treatment is most often caused by mutations in the
kinase domain of BCR-ABL that interfere with imatinib binding.
Dasatinib, approved in
mid-2006, is an orally available ABL kinase inhibitor that differs
from imatinib in that it can bind to both the active and inactive
conformations of the ABL kinase domain (Figure). Dasatinib
also inhibits a distinct spectrum of kinases that overlaps with the
array of enzymes that imatinib inhibits. Since it has less
stringent binding requirements than those of imatinib, dasatinib
has activity against many imatinib-resistant kinase domain
mutations of BCR-ABL at nanomolar concentrations. By targeting these kinases,
dasatinib inhibits the overproduction of leukemia cells in the bone marrow of
patients with CML and Ph+ALL and allows normal red cell, white cell, and blood
platelet production to resume. In cell-line models of CML and ALL, dasatinib
inhibited 18 of 19 imatinib-resistant BCR-ABL mutations within
a narrow concentration range, similar to that required to block wild-type
BCR-ABL. The only exception is a single mutation deep within the
ATP-binding pocket of the ABL tyrosine kinase (T315I) that confers
a high degree of resistance to imatinib and dasatinib and to the
imatinib analogue AMN-107, presumably as a result of steric hindrance caused
by replacement of threoninewith the bulkier isoleucine residue.
Pharmacokinetics:
11 Maximum plasma concentrations (Cmax) of dasatinib are observed
between 0.5 and six hours (Tmax) following oral administration. Dasatinib
exhibits dose-proportional increases in AUC and linear elimination
characteristics over the dosage range of 15 to 240 mg/day. While consumption
of a high-fat meal can result in a modest increase (14%) in the mean AUC, this
effect is not believed to be clinically relevant.
Dasatinib has an apparent
volume of distribution of 2,505 L, suggesting that the drug is extensively
distributed in the extravascular space. Binding of dasatinib and its active
metabolite (see below) to human plasma proteins in vitro was
approximately 96% and 93%, respectively, with no concentration dependence over
the range of 100 to 500 ng/mL.
Dasatinib is extensively
metabolized in humans, primarily by CYP3A4, resulting in formation of the
active metabolite. In human liver microsome assays, dasatinib is a weak,
time-dependent inhibitor of CYP3A4. The exposure of the active metabolite,
which is equipotent to dasatinib, represents only about 5% of the dasatinib
AUC, suggesting that this metabolite is unlikely to have a major role in the
drug's therapeutic profile. Other dasatinib metabolites are formed by the
action of flavin-containing monooxygenase 3 (FMO-3) and uridine
diphosphate-glucuronosyl-transferase (UGT) enzymes, and these metabolites
appear to be pharmacologically inactive.
Approximately 4% and 85% of
the administered dose of dasatinib is recovered in the urine and feces,
respectively, within 10 days. The parent drug accounts for 0.1% and 19% of the
administered dose in urine and feces, respectively, with metabolites being the
remainder of the dose. The overall mean terminal half-life of dasatinib is
three to five hours. In adults, the pharmacokinetics of dasatinib are not
significantly altered by age or gender. The pharmacokinetics of dasatinib have
not been evaluated in pediatric patients. In addition, no clinical studies
have been performed with dasatinib in patients with impaired hepatic or renal
function. Since less than 4% of dasatinib and its metabolites are excreted via
the kidney, renal function is not expected to significantly influence drug
exposure.
Clinical Profile:11,12
FDA approval of dasatinib was based on four single-arm, multicenter studies
that investigated the safety and efficacy of the drug in the treatment of
imatinib-resistant or imatinib-intolerant CML and Ph+ALL. These trials were
all ongoing at the time of approval. In all four trials, subjects received 70
mg of dasatinib twice daily. Each study investigated the drug in the treatment
of a single disease subclass (see below).
A chronic-phase CML study
enrolled 186 patients with imatinib-resistant (68%) or -intolerant (32%)
chronic-phase CML. Patients received treatment for a median of 5.6 months
(ranging from 0.03 to 8.3 months). Results yielded efficacy in the trial's
primary end point, with dasatinib producing major cytogenetic response (MCyR)
in 45% of subjects, including complete cytogenic response (CCyR, defined as 0%
Ph+ cells detected) in 33% of subjects. Furthermore, complete hematologic
response (CHR) was achieved in 90% of subjects.
An accelerated-phase CML study
enrolled 107 patients with imatinib-resistant (93%) or -intolerant (7%)
accelerated-phase CML. These patients received treatment for a median of 5.5
months (range, 0.2 to 10.1 months). Results yielded efficacy in the
trialprimary end point, with dasatinib producing major hematologic response
(MaHR) in 59% of subjects, including CHR in 33% and no evidence of leukemia
(NEL) in 26%. In addition, MCyR was achieved in 31% of subjects, including
CCyR in 21%.
The myeloid blast–phase CML
trial enrolled 74 patients with imatinib-resistant (92%) or intolerant (8%)
myeloid blast–phase CML. Subjects received therapy for a median of 3.5 months
(range, 0.03 to 9.2 months). Results yielded efficacy in the trial's primary
end point, with dasatinib producing MaHR in 32% of subjects, including CHR in
24% and NEL in 8%. In addition, MCyR was achieved in 30% of subjects,
including CCyR in 27%.
The lymphoid blast–phase CML
study enrolled 42 patients with imatinib-resistant (92%) or -intolerant (8%)
lymphoid blast–phase CML, and 36 patients with imatinib-resistant (94%) or
-intolerant (6%) Ph+ALL. CML subjects received treatment for a median of 2.8
months (range, 0.1 to 6.4 months), and Ph+ALL subjects received treatment for
a median of 3.2 months (range, 0.2 to 8.1 months). Dasatinib produced MaHR in
31% of CML subjects and 42% of Ph+ ALL subjects, including CHR in 26% and 31%
of subjects and NEL in 5% and 11% of subjects, respectively. In addition, MCyR
was achieved in 50% and 58% of subjects, including CCyR in 58% and 58% of
subjects, respectively.
Adverse Reactions:11,12
In clinical trials to date that involve 911 patients with leukemia (one phase
I and five phase II clinical studies), the majority of dasatinib-treated
patients experienced adverse drug reactions. The drug was discontinued for
adverse reactions in 6% of patients in chronic-phase, 5% in accelerated-phase,
and 11% in myeloid blast–phase CML and in 6% in lymphoid blast–phase CML or
Ph+ALL. The most frequently reported adverse effects included fluid retention
events (e.g., pleural effusion), GI effects (diarrhea, nausea, abdominal pain,
and vomiting) and bleeding events. Fluid retention was severe in 9% of
patients, including pleural and pericardial effusions. Severe ascites,
generalized edema, and severe pulmonary edema were reported in 1% of trial
patients. Fluid retention was typically managed by supportive care measures
such as diuretics or short courses of steroids. Patients who develop symptoms
suggestive of pleural effusion (dyspnea or dry cough) should be evaluated by
chest x-ray. Severe pleural effusion may require oxygen therapy and
thoracentesis.
Treatment with dasatinib is
associated with severe chronic toxicity criteria grade 3/4 thrombocytopenia,
neutropenia, and anemia, which occur more frequently in patients with advanced
CML or Ph+ALL than in patients with chronic-phase CML. Myelosuppression was
reported in patients with normal baseline laboratory values, as well as in
patients with preexisting laboratory abnormalities. Complete blood counts
should be performed weekly for the first two months and then monthly
thereafter, or as clinically indicated. In clinical studies, myelosuppression
was managed by interruption, dosage reduction, or discontinuation of dasatinib
therapy. Hematopoietic growth factor has been used in patients with persistent
myelosuppression.
Dasatinib-induced platelet
dysfunction and thrombocytopenia may result in severe hemorrhage. Severe GI
hemorrhage occurred in 7% of trial patients and generally required treatment
interruptions and transfusions. Severe central nervous system hemorrhage,
including fatalities, occurred in 1%. Other cases of severe hemorrhage
occurred in 4% of patients. Therefore, caution is advised when using dasatinib
in patients who are also required to take medications that inhibit platelet
function or anticoagulants.
In clinical trials, nine
patients receiving dasatinib had QTc prolongation as an adverse event.
Therefore, this drug should be administered with caution in patients who have
or may develop prolongation of QTc, such as patients with hypokalemia,
hypomagnesemia, or congenital long QT syndrome, as well as patients taking
antiarrhythmic drugs, other medicinal products that lead to QT prolongation,
or cumulative high-dose anthracycline therapy. Hypokalemia or hypomagnesemia
should be corrected prior to initiation of dasatinib therapy.
Grade 3/4 elevations of
transaminases or bilirubin were reported in all patients treated with
dasatinib, with increased frequency in patients with myeloid– or
lymphoid–blast CML or Ph+ALL. These elevations were managed with dose
reduction or interruption of therapy. Grade 3/4 hypocalcemia was reported in
patients with all phases of CML but with an increased frequency in patients
with myeloid– or lymphoid–blast CML or Ph+ALL. Patients with hypocalcemia
often had recovery with oral calcium supplementation.
Dasatinib is not recommended
for use in pregnant women or those contemplating pregnancy, since the drug may
cause fetal harm (pregnancy category D). Sexually active male or female
patients taking dasatinib should use adequate contraception.
Drug Interactions:11,12
Dasatinib is a substrate for CYP3A4. Therefore, drugs that inhibit this
isozyme (e.g., ketoconazole, itraconazole, erythromycin, cla
rithromycin, ritonavir, atazanavir, indinavir, nefazodone, nelfinavir,
saquinavir, and telithromycin) may increase dasatinib concentrations.
Concomitant use of such drugs with dasatinib should be avoided. If systemic
administration of a potent CYP3A4 inhibitor cannot be avoided, close
monitoring for toxicity and dosage reduction should be considered. Drugs that
induce CYP3A4 (e.g., dexamethasone, phenytoin, carbamazepine, and
phenobarbital) may decrease dasatinib concentrations. Alternative agents with
less enzyme-induction potential should be used, or a dosage increase of
dasatinib should be considered. St. John's wort (Hypericum perforatum
) may decrease dasatinib plasma concentrations unpredictably; thus, patients
taking dasatinib should not take St. John's wort.
In addition to functioning as
a substrate, dasatinib is a time-dependent inhibitor of CYP3A4. Therefore,
other drugs that are CYP3A4 substrates and have a narrow therapeutic index
(e.g., alfentanil, astemizole, terfenadine, cisapride, cyclosporine, fentanyl,
pimozide, quinidine, sirolimus, tacrolimus, or the ergot alkaloids ergotamine
and dihydroergotamine) should be administered with caution in patients treated
with dasatinib.
The solubility of dasatinib in the GI tract
is pH-dependent, and long-term suppression of gastric acid secretion by use of
H2 blockers (e.g., famotidine) or proton pump inhibitors (e.g.,
omeprazole) is likely to reduce dasatinib exposure. Concomitant use of H2
blockers or proton pump inhibitors with dasatinib is therefore not
recommended, and the use of antacids may be considered. If antacids are used,
they should be administered at least two hours before or after the dose of
dasatinib. Simultaneous administration of dasatinib and antacids should be
avoided.
Dosage and Administration:11
Dasatinib is available as 20, 50, and 70 mg white to off-white, biconvex,
round, film-coated tablets. The recommended dosage is 140 mg per day
administered orally in two divided doses (70 mg twice daily), one in the
morning and one in the evening with or without a meal. Tablets should not be
crushed or cut; they should be swallowed whole. A dosage increase or reduction
of 20-mg increments per dose is recommended based on individual safety and
tolerability.
CYP3A4 inducers such as
rifampin may decrease dasatinib plasma concentrations (see Drug Interactions
section). Selection of an alternate concomitant medication with no or minimal
enzyme-induction potential is recommended. If dasatinib must be administered
with a strong CYP3A4 inhibitor, a dosage decrease to 20 to 40 mg daily should
be considered. If the dosage of dasatinib is increased, the patient should be
monitored for toxicity.
In clinical studies of adult
patients with CML and Ph+ALL, dosage escalation to 90 mg twice daily
(chronic-phase CML) or 100 mg twice daily (advanced-phase CML and Ph+ALL) was
allowed in patients who did not achieve a hematologic or cytogenetic response
at the recommended dosage. In clinical studies, myelosuppression was managed
by dose interruption, dosage reduction, or discontinuation of therapy.
Hematopoietic growth factor has been used in patients with resistant
myelosuppression. Guidelines for dosage modifications are provided in the
manufacturer's literature.
If a severe nonhematologic
adverse reaction develops with dasatinib use, treatment must be withheld until
the event has resolved or improved. Treatment can then be resumed as
appropriate at a reduced dosage, depending on the initial severity of the
event.
Vorinostat (Zolinza, Merck)
Introduction and Mechanism
of Action:13-16 Non-Hodgkin's lymphomas (NHLs) are
subclassified into two grades based on growth rates. Low-grade lymphomas are
usually slow-growing, while high-grade lymphomas tend to grow more quickly.
Cutaneous T-cell lymphoma (CTCL) is a rare, low-grade lymphoma that accounts
for one in 20 of all NHL cases. It occurs most frequently in people between 40
and 60 years of age and affects up to 20,000 patients in the United States,
with another 1,500 new cases reported each year. Unlike other forms of NHL,
CTCL mainly affects the skin. It is caused by the uncontrolled growth of T
cells. Normal T cells function by regulating immune response to infection and
other foreign antigens. In CTCL, the malignant T cells accumulate and are
deposited in the skin.The most common subtypes of CTCL are mycosis fungoides
(MF) and Sézary syndrome (SS). CTCL is considered SS when large areas of the
skin are affected and large numbers of abnormal lymphocytes (Sézary cells) are
also found in the blood and lymph nodes. In some patients, there are no
plaques or tumors, but the whole skin can be red, thickened, swollen, and sore
(erythroderma). MF represents those forms of CTCL when the blood is not
affected.
Clinically, CTCL is staged
based on the degree of skin, lymph node, and visceral tissue involvement and
the presence of circulating Sézary cells, tumors, or erythroderma. Using this
system, CTCL is classified as: (1) stage Ia or limited patch/plaque MF
involving <10 % body surface area; (2) stage Ib or generalized patch/plaque MF
involving ?10% body surface area; (3) stage IIb or tumor stage MF; and
(4) stage III or erythrodermic MF/SS. In the early stage, small, raised, red
patches appear on the skin, commonly on the breast and buttocks, although they
can appear anywhere. At this stage, the disease often looks like a common skin
condition such as eczema or psoriasis. In the plaque or infiltrative stages,
irregularly shaped red patches (plaques) form. Although any part of the body
may be affected, the buttocks, skin folds, and face are particularly common
locations. There may be permanent hair loss from the affected areas if the
plaques are left untreated. In the tumor stage, raised tumors appear on the
skin and may become deep sores (ulcerate). At this stage, the cancer may have
also affected the lymph nodes and, rarely, internal organs such as the liver,
lungs, and spleen. CTCL does not necessarily progress through all three
stages, and only a small proportion of patients progress to this stage; most
never progress beyond the first stage.
A number of treatments can be
used for CTCL; therapy selection often depends on the stage or extent of skin
involvement. Although treatment for early or localized patch-stage MF may
result in a cure, the practical aim of therapy is generally to achieve and
maintain clinical remission, decrease morbidity, and palliate advanced
disease. Therapeutic modalities include topical therapy, phototherapy,
photopheresis (extracorporeal photochemotherapy), radiation therapy,
immunotherapy, chemotherapy, or newer agents such as antitumor vaccines and
antibody fusion toxins. Commonly used topical agents include high-potency
topical cortico steroids, carmustine, and mechlorethamine (nitrogen
mustard). Phototherapy may consist of psoralen with ultraviolet A
photochemotherapy (PUVA), ultraviolet-B (UVB) broadband (280 to 320 nm), and,
more recently, narrowband (TL-01-311 nm) UVB. Electron beam radiation has been
used locally and for total-body irradiation. Systemic drug therapies include
interferons (mostly alpha-interferon), retinoids, methotrexate, and other
drugs. Photopheresis has been employed for erythrodermic MF or SS. All of
these treatments can be used as monotherapy, and some have been used in
combination or in sequence.
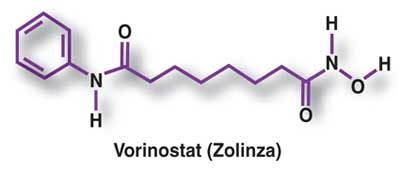
Vorinostat was approved by the
FDA in 2006 as a new therapy for cutaneous manifestations of CTCL in patients
who have progressive, persistent, or recurrent disease on or following two
systemic therapies (Figure). This drug has a novel mechanism of action,
functioning as an inhibitor of a number of histone deacetylase (HDAC)
isozymes. The HDACs catalyze the removal of acetyl groups from the lysine
residues of proteins, including histones and transcription factors. In some
cancer cells, there is an overexpression of HDACs or an aberrant recruitment
of HDACs to oncogenic transcription factors, causing hypoacetylation of core
nucleo somal histones. Hypoacetylation of histones is associated with a
condensed chromatin structure and repression of gene transcription. Inhibition
of HDAC activity allows for the accumulation of acetyl groups on the histone
lysine residues, resulting in an open chromatin structure and transcriptional
activation. In vitro, vorinostat inhibits the enzymatic activity of histone
deacetylase isozymes HDAC1, HDAC2, HDAC3 (class I), and HDAC6 (class II) at
nanomolar concentrations (IC50 <86 nM) and causes the accumulation
of acetylated histones. This is believed to induce cell cycle arrest and/or
apoptosis of some transformed cells.
Vorinostat will be made
accessible to patients through Merck's Accessing Coverage Today (ACT) program.
ACT is a three-part program designed specifically to assist patients in
obtaining vorinostat, offer help with insurance reimbursement issues, and
provide support for qualified individuals who lack insurance coverage for the
drug. Patients without coverage may be eligible for Merck's Patient Assistance
Program, which allows them to receive vorinostat free of charge. Merck is also
contributing to foundations that provide copay assistance to qualified
individuals.
Pharmacokinetics:16
In clinical trials, oral administration of a single 400-mg dose of vorinostat
with a high-fat meal, resulted in peak serum concentrations (Cmax) of 1.2 ±
0.62 µM, median Tmax of 4 (2 to 10) hours, and AUC of 5.5 ± 1.8 µM•hr. In the
fasting state, there was a modest decrease in the extent of absorption (4.2 ±
1.9 µM•hr) and increase in the rate of absorption (Tmax 1.5 hours), but these
differences are not expected to be clinically significant. At steady state in
the fed-state, oral administration of multiple 400-mg doses of vorinostat
resulted in a mean AUC and Cmax and a median Tmax of 6.0 ± 2.0
µM•hr and 1.2 ± 0.53 µM and 4 (0.5 to 14) hours, respectively. Vorinostat is
approximately 71% bound to human plasma proteins over the concentration range
of 0.5 to 50 µg/mL.
In vitro studies using human
liver microsomes indicate negligible biotransformation of vorinostat by CYP450
isozymes. Instead, vorinostat is metabolized by direct O
-glucuronidation and hydrolysis, followed by beta-oxidation, to yield
4-anilino-4-oxobutanoic acid. Both metabolites are pharmacologically inactive.
Compared to the parent drug, the mean steady-state serum exposures in humans
of the O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid
metabolite are fourfold and 13-fold higher, respectively. Less than 1% of the
oral dose of vorinostat is recovered in the urine unchanged, indicating that
renal excretion does not have a role in the elimination of this drug. The mean
urinary recovery of two pharmacologically inactive metabolites at steady state
was approximately 16% of the dose as the O-glucuronide of vorinostat
and 36% of the dose as 4-anilino-4-oxobutanoic acid. The total urinary
recovery of vorinostat and these two metabolites averaged 52 ± 13.3% of the
dose. The mean terminal half-life was approximately two hours for both
vorinostat and the O-glucuronide metabolite, while that of the
4-anilino-4-oxobutanoic acid metabolite was 11 hours.
The pharmacokinetics of
vorinostat do not appear to be vary significantly based on gender, race, or
age. Vorinostat was not evaluated in patients younger than 18 years or in
patients with hepatic or renal impairment. However, renal excretion does not
have an impact in the elimination of vorinostat.
Clinical Profile:16-18
The efficacy of vorinostat in the treatment of CTCL was assessed in two
open-label studies (study 1 and study 2) involving 107 patients. In both
studies, patients were treated until disease progression or intolerable
toxicity. In study 1, patients with advanced CTCL that was progressive,
persistent, or recurrent during or following two systemic therapies were
treated with vorinostat 400 mg once daily. The median age of patients was 60
years (51% male, 49% female). Approximately 18% of patients had stage Ib or
IIa CTCL, and 82% had stage IIb and higher CTCL. The primary end point was
response rate as determined by a modified Severity Weighted Assessment Tool
(SWAT) measuring the percentage of total-body surface area involvement.
Efficacy was measured as a complete clinical response (CCR), defined as no
evidence of disease, or a partial response, defined as a ?50% decrease
in SWAT skin assessment score compared with baseline. The overall objective
response rate was 29.7% in all patients treated with vorinostat. In patients
with stage IIb and higher CTCL, the overall objective response rate was 29.5%,
and one patient with stage IIb CTCL achieved a CCR. Secondary endpoints in
this study included time to objective response, time to progression, and
duration of objective response. In the study, the median time to response was
less than two months (55 days) in all patients. However, in rare cases, it
took up to six months for patients to achieve an objective response to
vorinostat. The median duration of response was not reached, since the
majority of responses continued at the time of analysis but was estimated to
exceed six months in all patients. The median time to progression approached
five months (148 days) in all patients, based on a criterion for tumor
progression of a 25% increase in SWAT score from the nadir.
In study 2, patients with CTCL
who were refractory or intolerant to one or more treatments were assigned to
one of three dosing cohorts. Patients in cohort 1 received 400 mg once daily,
while those in cohort 2 received 300 mg twice daily three days a week, and
those in cohort 3 received 300 mg twice daily for 14 days, followed by a
seven-day rest. Patients in cohort 3 received a maintenance regimen of 200 mg
twice daily during the rest period if no response was observed after 14 days
of full dosing. The primary efficacy end point in the study was objective
response, as measured by the seven-point Physician's Global Assessment scale.
In all patients treated, the objective response was 24.2% in the overall
population, 25% in patients with stage IIb or higher, and 36.4% in patients
with SS. The overall response rates were 30.8%, 9.1%, and 33.3% in cohorts 1,
2, and 3, respectively.
Adverse Reactions:16-18
In the two single-arm clinical studies described above, the most common side
effects, regardless of causality, included fatigue (52%), diarrhea (52%),
nausea (41%), taste alteration (28%), low platelet count (26%), anorexia
(24%), weight loss (21%), and muscle spasms (20%). The most common serious
adverse events, also regardless of causality, were pulmonary embolism (4.7%),
squamous cell carcinoma (3.5%), and anemia (2.3%). The GI adverse effects may
require management with antiemetic and antidiarrheal medications. Preexisting
nausea, vomiting, and diarrhea should be adequately controlled before
initiating therapy. Furthermore, because of the risk of dehydration from GI
effects, patients should be instructed to drink as least 2 L of fluid per day
to maintain adequate hydration during vorinostat therapy.
Since
treatment with vorinostat can cause dose-related thrombocytopenia and anemia,
platelet counts and hemoglobin should be monitored closely. If platelet counts
and/or hemoglobin are reduced during treatment, the dose of vorinostat should
be reduced or therapy should be discontinued. In addition, since pulmonary
embolism and deep vein thrombosis have been reported in patients taking
vorinostat, health care providers should be alert to the signs and symptoms of
these events, particularly in patients with a prior history of thromboembolic
events.
Serum glucose levels should be
monitored, especially in diabetic or potentially diabetic patients, because
hyperglycemia has been observed in some patients on vorinostat. Adjustment of
diet and/or therapy for hyperglycemia may be required. Baseline electrolytes
and ECGs should be determined, and these parameters should be monitored
periodically during vorinostat treatment, since this drug has been associated
with QTc prolongation and electrolyte abnormalities. Any electrolyte
abnormality (e.g., hypokalemia or hypomagnesemia) should be corrected prior to
initiation of vorinostat therapy.
While there are no adequate
controlled studies of vorinostat in pregnant women, animal studies suggest
that this drug has the potential to cause harm to the fetus. Therefore, if
vorinostat is used during pregnancy, or if the patient becomes pregnant while
taking vorinostat, the patient should be apprised of the potential hazard to
the fetus (pregnancy category D).
Drug Interactions:16-18
Vorinostat is not metabolized via the CYP isozymes and does not appear to
inhibit or induce these enzymes at therapeutic concentrations. Therefore,
vorinostat is not expected to interact when coadministered with drugs that are
known CYP substrates, inhibitors, or inducers. However, no formal clinical
studies have been conducted to evaluate potential cytochrome-based drug
interactions with vorinostat.
Prolongation of prothrombin
time and international normalized ratio were observed in patients receiving
vorinostat concomitantly with coumarin-derivative anticoagulants. Thus,
prothrombin time and international normalized ratio should be carefully
monitored in patients using vorinostat and coumarin concurrently. In addition,
severe thrombocytopenia and GI bleeding have been reported with concomitant
use of vorinostat and other HDAC inhibitors, such as valproic acid. Platelet
counts should be monitored every two weeks for the first two months of
therapy.
Dosing and Administration:
16 Vorinostat is supplied as 100 mg white, opaque, hard gelatin
capsules. These capsules should not be opened or crushed. The recommended
dosage is 400 mg once daily with food. If a patient is intolerant to therapy,
the dosage may be reduced to 300 mg once daily with food. The dosage may be
further reduced to 300 mg once daily with food for five consecutive days each
week, as necessary. No information is available in patients with renal or he
patic impairment. However, since vorinostat is cleared primarily by
metabolism, patients with hepatic impairment should be treated with caution.
Treatment may be continued as long as there is no evidence of progressive
disease or unacceptable toxicity.
References
1. Ellepola ANB,
Samaranayake LP. Antimycotic agents in oral candidosis: an overview: clinical
variants. Dent Update. 2000;27:111–116.
2. Rex JH, Rinald MG,
Pfaler MA. Resistance of Candida species to fluconazole. Antimicrob Agents
Chemother. 1995;39:1-8.
3. NOXAFIL®
(posaconazole) Oral Suspension, Product Information, Schering Corporation,
Kenilworth, NJ, 2006.
4. Dodds Ashley ES,
Alexander BD. Posaconazole. Drugs Today. 2005;41:393-400.
5. Courtney R, Pai S,
Laughlin M, et al. Pharmacokinetics, safety, and tolerability of oral
posaconazole administered in single and multiple doses in healthy adults.
Antimicrob Agents Chemother. 2003;47:2788-2795.
6. Groll AH, Walsh TJ.
Antifungal efficacy and pharmacodynamics of posaconazole in experimental
models of invasive fungal infections. Mycoses.2006;49(suppl 1):7-16.
7. Notheis G, Tarani L, Costantino F, et al. Posaconazole for treatment of
refractory invasive fungal disease. Mycoses. 2006;49(suppl 1):37-41.
8. Carrillo-Munoz AJ, Quindos
G, Ruesga M, et al. Antifungal activity of posaconazole compared with
fluconazole and amphotericin B against yeasts from oropharyngeal candidiasis
and other infections. Journal of Antimicrobial Chemotherapy.
2005;55:317-319.
9. Bradeen HA, Eide CA, O'Hare T, et al. Comparison of imatinib, dasatinib
(BMS-354825), and nilotinib (AMN107) in an n-ethyl-n-nitrosourea (ENU)-based
mutagenesis screen: high efficacy of drug combinations. Blood2006; Jun
13.
10. Tokarski JS, Newitt JA, Chang CY, et al. The structure of Dasatinib
(BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory
activity against imatinib-resistant ABL mutants. Cancer Res.
2006;66:5790-5797.
11. SPRYCEL™ (dasatinib) Tablets, Product Information, Bristol-Myers Squibb
Company, Princeton, NJ, June 2006.
12. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant
Philadelphia chromosome-positive leukemias. New Engl J Med.
2006;354:2531-2541.
13. Siegel RS, Pandolfino T, Guitart J, et al. Primary cutaneous T-cell
lymphoma: review and current concepts. J Clin Oncol. 2000;18:2908-2925.
14. Scarisbrick J. Treatment of mycosis fungoides and Sézary syndrome: recent
advances and novel therapies. Expert Rev Dermatol. 2006;1:569-577.
15. Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides.
N Engl J Med. 2004;350:1978-1988.
16. ZOLINZA™ (vorinostat) Capsules, Product Information, Merck & Co. Inc.,
Whitehouse Station, NJ, October 2006.
17. Olsen E, Kim YH, Kuzel T, et al. Vorinostat (suberoyl anilide
hydroxamic acid, SAHA) is clinically active in advanced cutaneous T-cell
lymphoma (CTLC): results of a phase IIb trial. J Clin Oncol.
2006;24(suppl 18):A-7500.
18. O'Connor OA, et al. Clinical experience with intravenous and oral
formulations of the novel histone deacetylase inhibitor suberoylanilide
hydroxamic acid in patients with advanced hematologic malignancies. J Clin
Oncol. 2006;24:166-173.



