US Pharm. 2010;35(10):HS2-HS18.
New molecular entities (NMEs), as defined by the FDA, are drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2009–2010 (TABLE 1) detail the basic clinical and pharmacologic profiles for each new drug, as well as its pharmacokinetics, adverse reactions, drug interactions, and dosing data. Note that the information for each NME was obtained primarily from sources published prior to FDA approval; thus, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature, such as the emergence of additional adverse reactions and black box warnings.
Dalfampridine (Ampyra, Acorda Therapeutics)
Indication and Clinical Profile1,2: Dalfampridine is the first and only FDA-approved oral drug as a treatment to improve walking in patients with multiple sclerosis (MS). MS is a chronic, often disabling, disease that affects the brain, spinal cord, and optic nerves. There are about 400,000 people in the United States and 2.5 million people worldwide with MS. The progress, severity, and specific symptoms of MS are variable and unpredictable from person to person. Symptoms can be mild (as numbness in the limbs) or more severe and include paralysis or loss of vision. Typically within 15 years of diagnosis, 50% of patients with MS will require assistance walking. A significant number of patients with MS also experience cognitive impairment (i.e., difficulties in concentration, attention, memory, and judgment) and depression.
Dalfampridine may be given in conjunction with any FDA-approved disease-modifying therapy, including interferons, glatiramer acetate, mitoxantrone, and natalizumab.
The efficacy of dalfampridine was demonstrated in two phase III clinical trials in which the primary end point was walking speed (in feet per second) in a timed 25-foot walk (T25FW). A significantly greater proportion of patients taking dalfampridine 10 mg twice daily showed faster walking speeds for at least three visits out of a possible four compared with patients taking placebo (T25FW responder rate in Trial 1: 34.8% vs. 8.3%; Trial 2: 42.9% vs. 9.3%). The increased T25FW response rate in the dalfampridine group was seen regardless of the type of MS. Moreover, in both trials, consistent improvements in T25FW were shown to be associated with improvements on the 12-item Multiple Sclerosis Walking Scale (MSWS-12), a validated patient self-assessment of ambulatory disability.
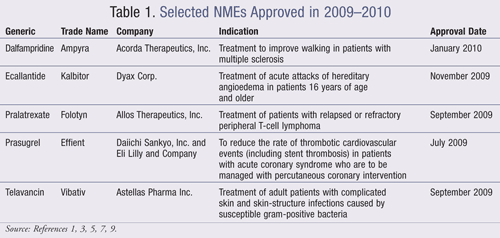
Pharmacology and Pharmacokinetics1: Dalfampridine is a 4-aminopyridine (FIGURE 1), which is a broad-spectrum potassium channel blocker that can produce increased action potential conduction in demyelinated nerves. It remains to be determined whether these actions are directly related to its therapeutic efficacy in MS.
The extended-release oral formulation of dalfampridine is well absorbed from the gastrointestinal (GI) tract (>90%), producing peak plasma levels in 3 to 4 hours. Food does not alter absorption. Dalfampridine is excreted primarily in the urine as the parent drug (>90%). The two known minor metabolites, 3-hydroxy-4-aminopyridine (4%) and 3-hydroxy-4-aminopyridine sulfate (3%), have no pharmacologic activity. The elimination half-life of dalfampridine is 5 to 7 hours.
Adverse Reactions and Drug Interactions1,2: In clinical trials, the most common adverse reactions in patients treated with dalfampridine included urinary tract infection, insomnia, dizziness, headache, nausea, weakness, back pain, balance disorder, swelling in the nose or throat, constipation, diarrhea, indigestion, throat pain, and burning, tingling, or itching of the skin. At doses >10 mg bid, dalfampridine can causes seizures. Thus, it is contraindicated in patients with a prior history of seizure and should be discontinued in patients in which seizure occurs. It also should not be used in patients with moderate-to-severe kidney disease, since plasma levels may increase and approach those associated with the occurrence of seizures.
To date, no clinically significant drug interactions have been identified, and the drug does not prolong the QTc interval or have a clinically important effect on QRS duration. Dalfampridine is a Pregnancy Category C drug, and its safety in breastfeeding is not known.
Dosage and Administration1: Dalfampridine is supplied as a 10-mg, film-coated, extended-release tablet. The maximum recommended dose is 10 mg twice daily, approximately 12 hours apart with or without food. No additional benefit was demonstrated at doses >10 mg twice daily, and adverse events, including seizures, were more frequent at higher doses. Tablets should be taken whole and not divided, crushed, chewed, or dissolved. Patients should not take double or extra doses if a dose is missed. Dalfampridine is contraindicated in patients with moderate-to-severe renal impairment since plasma levels may rise to those associated with an increased risk of seizures.
Ecallantide (Kalbitor, Dyax Corp.)
Indication and Clinical Profile3,4: Ecallantide was approved by the FDA to treat hereditary angioedema (HAE), an acute inflammatory condition characterized by episodes of severe, often painful swelling affecting the extremities, GI tract, genitalia, and larynx. HAE attacks, which occur more than 20 times annually, are unpredictable and range in progression and severity. Furthermore, they may lead to permanent disfigurement, disability, or death. HAE is a rare genetic condition caused by a mutation in the C1 esterase-inhibitor (C1-INH) gene, resulting in low levels of this blood protein. C1-INH plays a role in regulating the immune system and intrinsic clotting pathways. It is estimated that 10,000 people in the U.S. have HAE. Ecallantide is the second drug marketed in the U.S. to treat HAE attacks; earlier, Berinert (a human blood–derived C1-INH) was approved to treat facial and abdominal attacks of HAE.
The safety and efficacy of ecallantide were evaluated in two controlled trials known as EDEMA3 and EDEMA4. Patients having an attack of HAE, at any anatomical location (abdominal, peripheral, or laryngeal), and having at least one moderate or severe symptom were treated with 30-mg subcutaneous (SC) injections of ecallantide or placebo. Because patients could participate in both trials, a total of 143 unique patients participated. There were 64 patients with abdominal attacks, 55 with peripheral attacks, and 24 with laryngeal attacks. In both trials, the effects of ecallantide were evaluated using the Mean Symptom Complex Severity (MSCS) score and the Treatment Outcome Score (TOS). These measures evaluated the severity of attack symptoms at all anatomical locations (MSCS score) and response to therapy (TOS).
In the EDEMA4 trial at 4 hours, patients treated with ecallantide demonstrated a greater decrease from baseline in the mean MSCS than placebo (-0.8 vs. -0.4) and a greater mean TOS (53 vs. 8). At 24 hours, patients treated with ecallantide also demonstrated a greater decrease from baseline in the mean MSCS than placebo (-1.5 vs. -1.1) and a greater mean TOS (89 vs. 55).
Pharmacology and Pharmacokinetics3: HAE is a rare genetic disorder caused by mutations to C1-INH located on chromosome 11q. It is inherited as an autosomal dominant trait. HAE is characterized by low levels of C1-INH activity and low levels of C4. C1-INH functions to regulate the activation of the complement and intrinsic coagulation (contact system pathway) and is a major endogenous inhibitor of plasma kallikrein. The kallikrein-kinin system is a complex proteolytic cascade involved in the initiation of both inflammatory and coagulation pathways. One critical aspect of this pathway is the conversion of high-molecular-weight (HMW) kininogen to bradykinin by the protease plasma kallikrein, a pathway inhibited endogenously by C1-INH. In HAE, normal regulation of plasma kallikrein activity and the classical complement cascade is not present. During attacks, unregulated activity of plasma kallikrein results in excessive bradykinin generation. Bradykinin is a vasodilator believed to be responsible for the characteristic HAE symptoms of localized swelling, inflammation, and pain. Ecallantide is a potent (Ki = 25 pM), selective, and reversible inhibitor of human plasma kallikrein. By blocking the kallikrein binding site, this drug inhibits the conversion of HMW kininogen to bradykinin.
Maximum plasma concentrations (Cmax, 500-600 ng/mL) following administration of a single 30-mg SC dose of ecallantide are observed within 2 to 3 hours. The mean area under the concentration-time curve (AUC) was about 3,017 ng*h/mL. Following administration, plasma concentration declined with a mean elimination half-life of 2.0 hours. Plasma clearance was 153 mL/min and the volume of distribution was 26.4 L. Ecallantide is a small protein (7,054 daltons) that appears to be cleared renally. Based on a population pharmacokinetic analysis, body weight, age, and gender were not found to affect ecallantide exposure significantly. No pharmacokinetic data are available in patients or subjects with hepatic or renal impairment.
Adverse Reactions and Drug Interactions3,4: In clinical trials to date involving 255 HAE patients treated with ecallantide, 10 patients (3.9%) experienced anaphylaxis. Anaphylaxis occurred most commonly within the first hour after injection of ecallantide, and symptoms associated with these reactions included chest discomfort, flushing, pharyngeal edema, pruritus, rhinorrhea, sneezing, nasal congestion, throat irritation, urticaria, wheezing, and hypotension. Because of the risk of anaphylaxis, ecallantide should only be administered by a health care professional, in a medical setting where serious anaphylactic reactions and HAE can be treated (black box warning). Ecallantide should also not be administered to patients with known clinical hypersensitivity to the drug. Other common side effects of ecallantide use include headache, nausea, fatigue, diarrhea, upper respiratory tract infection, injection site reactions, nasopharyngitis, vomiting, pruritus, upper abdominal pain, and pyrexia. No formal drug-interaction studies have been performed to date.
Dosage and Administration3: Ecallantide is provided as a colorless liquid in 10 mg/mL vials. The recommended dose is 30 mg (3 mL), administered subcutaneously in three 10-mg (1-mL) injections. If attack persists, an additional dose of 30 mg may be administered within a 24-hour period. Ecallantide should only be administered by a health care professional with appropriate medical support to manage anaphylaxis and HAE. In addition, patients and health care providers can contact the Kalbitor Access program to receive information and work with program staff to research patient insurance coverage.
Pralatrexate (Folotyn, Allos Therapeutics)
Indication and Clinical Profile5,6: Pralatrexate is a chemotherapy agent indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). It was approved under the FDA’s accelerated approval process, which allows earlier approval of drugs for unmet medical needs (orphan designation).
The efficacy of pralatrexate was demonstrated in a phase II international clinical trial in 115 patients with PTCL who had relapsed or had progressive disease following prior therapy. The median number of prior systemic therapies was 3 (range 1 to 12), and 24% of patients did not have an objective response to any previous therapies. Of the enrolled patients, 111 received pralatrexate at a starting dose of 30 mg/m2 IV once weekly for 6 weeks followed by a 1-week break (1 cycle). In addition, each patient received vitamin B12 (1.0 mg intramuscular [IM] injection) every 8 to 10 weeks and a daily administration of folic acid (1.0-1.25 mg orally). Patients who had tumor responses or stable disease continued to receive additional cycles until disease progression or unacceptable toxicity. Imaging scans were performed to assess disease status at week 7 (end of cycle 1) and subsequently at 14-week intervals.
Overall, approximately 27% of patients experienced a complete (8.25%) or partial (18.35%) response to treatment. The median response duration was 9.4 months (range 1-503 days) and 13 patients (12%) had response durations of at least 14 weeks.
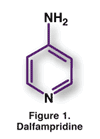
Pharmacology and Pharmacokinetics5: Pralatrexate, an analogue of methotrexate (FIGURE 2), is mixture of R- and S-diastereomeric folate derivatives that competitively inhibit dihydrofolate reductase and prevent formation of essential folate cofactors. These isomers also competitively inhibit polyglutamylation by the enzyme folylpolyglutamyl synthetase. By inhibition of these processes, pralatrexate isomers block the single carbon transfer reactions necessary for the construction of thymidine and purines, precursors required for the biosynthesis of DNA.
Pralatrexate Cmax and total systemic exposure (AUC) increase proportionally with standard IV dosing, with a total systemic clearance of 417 mL/min for the S-diastereomer and 191 mL/min for the R-diastereomer. Pralatrexate is not a substrate or inhibitor of p-glycoprotein transporters or cytochrome enzymes. The mean fraction of unchanged pralatrexate diastereomers excreted in urine is 31% for the S-diastereomer and 38% for the R-diastereomer. The terminal elimination half-life of pralatrexate is 12 to 18 hours.
Adverse Reactions5,6: In clinical trials with pralatrexate, mucositis, thrombocytopenia, nausea, fatigue, anemia, constipation, pyrexia, edema, cough, epistaxis, vomiting, neutropenia, and diarrhea were the most common adverse reactions (>3%). Adverse reactions were the reason for dose reductions in 31% of patients, dose omission in 69%, and treatment withdrawal in 23%. Overall, 85% of scheduled doses were administered. Eight deaths were reported within 30 days of the last pralatrexate dose; seven were attributed to progressive disease and one was due to cardiopulmonary arrest possibly related to the drug. Pralatrexate is classified as a Pregnancy Category D drug because it has the potential to cause fetal harm when administered to a pregnant woman.
Drug Interactions5: No formal clinical assessments of pharmacokinetic drug–drug interactions between pralatrexate and other drugs have been conducted. Data from in vitro studies suggest pralatrexate is not a substrate, inhibitor, or inducer of CYP450 isoenzymes and thus has low potential for interactions with other drugs metabolized by these enzymes. However, due to the contribution of renal excretion to the overall clearance of pralatrexate (~34%), concurrent administration of other drugs subject to substantial renal clearance (e.g., probenecid, nonsteroidal anti-inflammatory drugs [NSAIDs], trimethoprim/sulfamethoxazole) may result in delayed clearance of pralatrexate.
Dosage and Administration5: Pralatrexate is supplied as single-use vials containing 20 mg/mL of the drug in 1 mL solution in a vial (20 mg/1 mL) and 2 mL solution in a vial (40 mg/2 mL). The recommended dose of pralatrexate is 30 mg/m2 administered as an IV push over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles until progressive disease or unacceptable toxicity. Patients must be supplemented with vitamin B12, 1 mg IM every 8 to 10 weeks, and folic acid 1.0 to 1.25 mg orally each day. Treatment interruption or dose reduction to 20 mg/m2 may be needed to manage adverse drug reactions.
Prasugrel (Effient, Daiichi Sankyo and Eli Lilly)
Indication and Clinical Profile7,8: Prasugrel is a thienopyridine-class inhibitor of platelet activation and aggregation. This agent is approved to reduce the rate of thrombotic cardiovascular (CV) events in patients with acute coronary syndrome (ACS) who are undergoing percutaneous coronary intervention (PCI), including patients with unstable angina (UA) or non-ST-elevation myocardial infarction (NSTEMI) and patients with ST-elevation myocardial infarction (STEMI) managed with primary or delayed PCI.
The approval of prasugrel was based primarily on results obtained in the multicenter, international TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel) trial that compared prasugrel against clopidogrel in over 13,000 patients with ACS who were undergoing PCI. The loading dose of prasugrel (60 mg) or clopidogrel (300 mg) was administered between randomization and 1 hour after patients left the catheterization laboratory. Patients were then treated with prasugrel 10 mg/day or clopidogrel 75 mg/day and followed up for ≥6 months. The primary outcome was the composite of CV death, nonfatal MI, or nonfatal stroke. This end point occurred less frequently in prasugrel-treated patients than in clopidogrel-treated patients in both the UA/NSTEMI group and the STEMI group. In both groups, this reduction in the composite end point was driven by a reduction in nonfatal MI. Prasugrel, however, was associated with a higher rate of clinically significant bleeding than clopidogrel.
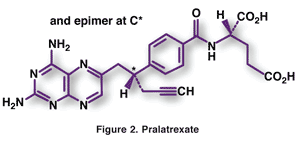
Pharmacology and Pharmacokinetics7: Prasugrel is an inhibitor of platelet activation and aggregation through the irreversible binding of its active metabolite to the P2Y12 class of platelet adenosine diphosphate (ADP) receptors (FIGURE 3). Prasugrel produces inhibition of platelet aggregation to 20 µM or 5 µM ADP, as measured by light transmission aggregometry. Following a 60-mg loading dose, approximately 90% of patients had at least 50% inhibition of platelet aggregation by 1 hour. Maximum platelet inhibition was about 80%. Mean steady-state inhibition of platelet aggregation was about 70%, following 3 to 5 days of dosing at 10 mg daily after a 60-mg loading dose of prasugrel.
Prasugrel is a prodrug that is well absorbed (80%) upon oral administration and rapidly metabolized to a pharmacologically active metabolite and inactive metabolites. The active metabolite is formed by hydrolysis to the thiolactone, followed by oxidation by CYP3A4, CYP2B6, and, to a lesser extent, CYP2C9 and CYP2C19. Peak plasma levels of the active metabolite occur 30 minutes after dosing. Coadministration with a high-fat meal increases the time to peak plasma concentration (tmax) and decreases Cmax levels, but does not alter the AUC. The active metabolite has an elimination half-life of about 7 hours (range 2-15 h). Dosage adjustment is not required in patients with renal impairment or mild-to-moderate hepatic insufficiency.
Adverse Reactions7,8: The most common adverse events associated with prasugrel treatment include bleeding, severe thrombocytopenia, anemia, abnormal hepatic function, allergic reactions, angioedema, hypertension, hypercholesterolemia/hyperlipidemia, and headache. Bleeding should be suspected in patients who are hypotensive and have recently undergone coronary angiography, PCI, coronary artery bypass grafting (CABG), or another surgical procedure, even if no overt signs of bleeding are present. Prasugrel is contraindicated in patients with active bleeding or a history of transient ischemic attack (TIA) or stroke (black box warning). Additional risk factors for bleeding include age ≥75 years, CABG or other surgical procedure, body weight <60 kg, propensity for bleeding, and medications that increase bleeding risk. Prasugrel should be discontinued ≥7 days before CABG when possible. Prasugrel should also be discontinued in the event of active bleeding, elective surgery, stroke, or TIA. Premature discontinuation of prasugrel is associated with increased risk of cardiac events; therefore, lapses in therapy should be avoided. Patients treated with other thienopyridines have experienced thrombotic thrombocytopenic purpura.
Drug Interactions7,8: Prasugrel can be administered with aspirin (75-325 mg/day), heparin, GPIIb/IIIa inhibitors, statins, digoxin, and drugs that elevate gastric pH, including proton pump inhibitors and H2-blockers. Coadministration with warfarin or NSAIDs increases the risk of bleeding. Prasugrel can also be administered with drugs that are inducers or inhibitors of CYP450 enzymes.
Dosage and Administration7: Prasugrel is supplied as 5- and 10-mg film-coated tablets. Therapy should be initiated as a single 60-mg oral loading dose and then continued at a dose of 10 mg once daily with or without food. Prasugrel-treated patients should also receive aspirin 75 to 325 mg/day. For patients who weigh <60 kg, health care professionals should consider lowering the maintenance dose of prasugrel to 5 mg/day.
Telavancin (Vibativ, Astellas Pharma)
Indication and Clinical Profile9,10: Telavancin is a structurally novel antimicrobial that has been approved for the treatment of complicated skin and skin-structure infections (cSSSI) caused by susceptible isolates of the gram-positive bacteria. The efficacy of this agent was assessed in two multinational trials that compared IV telavancin (10 mg/kg q24h) with IV vancomycin (1 g q12h) for 7 to 14 days. The primary efficacy end point was the clinical cure rate at follow-up. In the first trial, clinically evaluable patients in the telavancin treatment group had a clinical cure rate of 84% versus 83% for vancomycin-treated patients. In the second study, telavancin-treated patients had a clinical cure rate of 84% versus 88% among vancomycin-treated patients. A pooled analysis of these two trials for similar infections found telavancin to be noninferior to vancomycin. In addition, in two unpublished studies involving the treatment of gram-positive, hospital-acquired pneumonia, telavancin was found to be noninferior to vancomycin. Thus, whether telavancin will offer any benefit against organisms with reduced susceptibility to vancomycin in these or other infections remains to be established.
Pharmacology and Pharmacokinetics9: Telavancin is a lipoglycopeptide antibacterial agent that is a synthetic derivative of vancomycin. This antibiotic inhibits bacterial cell wall synthesis by interfering with the polymerization and cross-linking of peptidoglycan, and also binds to the bacterial membrane, disrupting its barrier function. In vitro, telavancin is bactericidal against common gram-positive pathogens including Staphylococcus aureus (including methicillin-resistant strains or MRSA), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus anginosus group, Streptococcus pneumoniae (including penicillin-resistant isolates), and Enterococcus faecalis (vancomycin-susceptible isolates only). Like vancomycin, it is ineffective against gram-negative pathogens.
Over typical IV dose ranges and treatment times, telavancin pharmacokinetics are linear, and steady-state concentrations are achieved by the third daily dose. Telavancin is highly bound (90%) to plasma proteins, primarily to serum albumin, and binding is not affected by renal or hepatic impairment. The drug appears to undergo a small degree of metabolism by noncytochrome enzymes, but the precise pathway of metabolism has not been determined. Telavancin has a relatively long half-life (7-9 h) as well as a prolonged postantibiotic effect (4-6 h). It is primarily eliminated by the kidney as the parent drug. Thus, dosage adjustment is required in patients with significant renal impairment.
Adverse Reactions9,10: The most common adverse reactions occurring in >10% of telavancin-treated patients in trials to date included taste disturbance, nausea, vomiting, and foamy urine. The infusion-related reaction known as red man syndrome, which occurs with vancomycin, might also occur with telavancin. Thus, this drug should be administered over a 60-minute period, as more rapid IV infusions can lead to upper body flushing, urticaria, pruritus, or rash. Renal dysfunction, usually reversible after drug discontinuation, occurred in 3% of patients treated with telavancin, compared to 1% of those treated with vancomycin. Patients with creatinine clearance (CrCl) ≤50 mL/min who were treated with telavancin demonstrated lower clinical cure rates than patients with CrCl >50 mL/min. Nearly all antibacterial agents have been associated with cases of Clostridium difficile–associated diarrhea, which may range from mild diarrhea to fatal colitis. Telavancin does not interfere with coagulation, but it does interfere with some tests used to assess coagulation. QTc prolongation has occurred in a small percentage of patients, but no associated cardiovascular adverse events or arrhythmias have been reported.
In animals, use of telavancin during pregnancy reduced fetal weights and increased rates of digit and limb deformities in offspring. Thus, women of childbearing potential should have a serum pregnancy test before treatment and should use effective contraception during treatment with telavancin. Pregnant women should avoid using telavancin unless the potential benefits outweigh the potential risks to the fetus (Pregnancy Category C).
Drug Interactions9: Telavancin is a CYP3A4 inhibitor in vitro, but in human trials it did not alter the pharmacokinetic profile of midazolam, a prototype CYP3A4 substrate. Caution is advised with use of telavancin in patients taking other QTc prolonging agents. Telavancin binds to the artificial phospholipid surfaces added to many anticoagulation tests, thereby interfering with assays including prothrombin time (PT), international normalized ratio (INR), and activated partial thromboplastin time (aPTT).
Dosage and Administration9: Telavancin is supplied as 250-mg and 750-mg single-dose vials. It should be administered as a 10-mg/kg dose through IV infusion over a 60-minute period once every 24 hours for 7 to 14 days. Therapy duration should be based on the severity and site of the infection and the patient’s clinical and bacteriological progress. In patients with CrCl of 30 to 50 mL/min, a 7.5-mg/kg dose should be administered every 24 hours; in patients with CrCl of 10 to ≤30 mL/min, a 10-mg/kg dose should be administered every 48 hours. At present, data are insufficient to make specific dosage adjustment recommendations for patients with a CrCl <10 mL/min. A pregnancy test and serum creatinine determination should be done before starting therapy with telavancin, and renal function should be monitored during therapy. Monitoring of serum concentrations is not currently recommended with telavancin as it is with vancomycin.
REFERENCES
1. Ampyra (dalfampridine) package insert. Hawthorne, NY: Acorda Therapeutics, Inc; January 2010.
2. Goodman AD, Brown TR, Krupp LB, et al; Fampridine MS-F203 Investigators. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;28:697-698.
3. Kalbitor (ecallantide) package insert. Cambridge, MA: Dyax Corp; November 2009.
4. Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104:523-529.
5. Folotyn (pralatrexate injection) package insert. Westminster, CO: Allos Therapeutics, Inc; September 2009.
6. O’Connor OA, Horwitz S, Hamlin P, et al. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol. 2009;27:4357-4364.
7. Effient (prasugrel) package insert. Indianapolis, IN: Daiichi Sankyo, Inc. and Eli Lilly and Company; July 2009.
8. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
9. Vibativ (telavancin for injection) package insert. Deerfield, IL: Astellas Pharma Inc; September 2009.
10. Stryjewski ME, Graham DR, Wilson SE, et al. Telavancin versus vancomycin for the treatment of complicated skin and skin-structure infections caused by gram-positive organisms. Clin Infect Dis. 2008;46:1683-1693.
To comment on this article, contact rdavidson@uspharmacist.com.



