US Pharm. 2014;39(10):HS2-HS14.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2013–2014 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
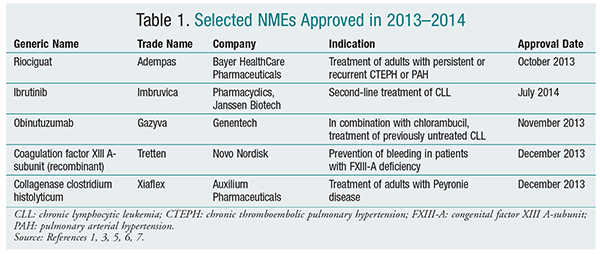
Riociguat (Adempas, Bayer HealthCare Pharmaceuticals)
Indication and Clinical Profile1,2: Riociguat was approved to treat persistent and recurrent chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH). Pulmonary hypertension is caused by abnormally high blood pressure in the arteries of the lungs that makes the right side of the heart work harder than normal. In its various forms, pulmonary hypertension is a chronic, progressive, debilitating disease, often leading to death or the need for lung transplantation.
Approval of riociguat was based on two multicenter, double-blind, randomized, placebo-controlled trials: the CHEST-1 trial for CTEPH and the PATENT-1 trial for PAH. Both trials used as a primary endpoint the change in 6-minute walking distance (6MWD) from baseline. CHEST-1 was conducted over 16 weeks in 261 patients with inoperable CTEPH, and PATENT-1 was conducted over 12 weeks in 445 patients with PAH that was either untreated or treated with a prostacyclin analogue or an endothelin receptor blocker. CHEST-1 and PATENT-1 demonstrated significant improvements in 6MWD of 46 min and 36 min, respectively, in patients treated with riociguat versus placebo. In CHEST-1, riociguat therapy also resulted in statistically significant improvements in pulmonary vascular resistance, N-terminal prohormone brain natriuretic peptide, and World Health Organization functional class.
Pharmacology and Pharmacokinetics1,2: Riociguat (FIGURE 1) is a diaminopyrimidine that acts on soluble guanylate cyclase (sGC), a protein that, upon binding with nitric oxide (NO), catalyzes the formation of guanosine monophosphate (cGMP). cGMP then acts as a second messenger, resulting in subsequent vasodilation in vascular smooth muscle in the cardiopulmonary system. Riociguat stabilizes NO binding to sGC and also acts as a direct stimulator of sGC independently of NO.
The absolute bioavailability of riociguat is 94% when the drug is taken with or without food. It reaches a peak plasma concentration in about 1.5 hours and has a half-life of 12 hours. Riociguat undergoes metabolism by several cytochrome isozymes, with the formation of the major metabolite being catalyzed by CYP1A1 followed by N-glucuronidation. It does not appear to significantly induce or inhibit metabolism by any major cytochrome isozyme. Riociguat is eliminated primarily both renally (40%) and fecally (53%).
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions (≥5%) reported in clinical trials were gastrointestinal abnormalities (dyspepsia/gastritis, nausea, diarrhea, vomiting, constipation, gastroesophageal reflux), headache, dizziness, edema, anemia, and hypotension. Serious bleeding events also occurred in 2.4% of patients taking riociguat. The drug’s label has a boxed warning stating that riociguat should not be given to women who are pregnant or may become pregnant, owing to its potential for embryo-fetal toxicity (Pregnancy Category X). Female patients can receive the drug only through enrollment in the Adempas Risk Evaluation and Mitigation Strategies (REMS) program. Females with reproductive potential also must comply with the program’s requirements for pregnancy testing and contraception (see manufacturer’s program guidelines). Physicians and pharmacies must enroll in the REMS restricted-distribution program in order to be certified to prescribe and dispense riociguat.
Riociguat should not be used concomitantly with nitrates, NO donors, or phosphodiesterase inhibitors, owing to the increased risk of hypotensive events. Coadministration with strong CYP inhibitors and P-glycoprotein (Pgp)/breast cancer resistance protein (BCRP) inhibitors should be avoided, as these will increase riociguat exposure and may also increase the risk of hypotensive events. As smoking has been shown to reduce riociguat concentrations by 50% to 60%, patients who smoke may require a dosage >2.5 mg per day to equal the drug exposure in nonsmokers.
Dosage and Administration1,2: Riociguat is supplied as 0.5-mg, 1-mg, 1.5-mg, 2-mg, and 2.5-mg tablets. The recommended starting dosage is 1 mg three times daily, and if the patient has no signs or symptoms of hypotension (systolic blood pressure >95 mmHg), the dosage may be up-titrated by 0.5 mg three times daily to a maximum of 2.5 mg three times daily. Dosage increases should be made no sooner than 2 weeks apart. The dosage should be decreased by 0.5 mg taken three times daily if the patient shows symptoms of hypotension. In patients who smoke, dosages higher than 2.5 mg three times daily may be considered; however, a reduction may be required upon smoking cessation. A starting dosage of 0.5 mg three times daily should be considered in patients who are also taking strong CYP or Pgp/BCRP inhibitors. No dosage adjustment is recommended for elderly patients or those with renal impairment or mild-to-moderate hepatic impairment. Riociguat is not recommended in patients with creatinine clearance <15 mL/min or in those on dialysis. The safety and efficacy of riociguat have not been established in pediatric patients or in patients with severe hepatic impairment.
Ibrutinib (Imbruvica, Pharmacyclics, Janssen Biotech)
Indication and Clinical Profile3,4: The FDA has approved ibrutinib for second-line treatment of chronic lymphocytic leukemia (CLL). Earlier, ibrutinib was approved as a second-line treatment for mantle cell lymphoma (MCL), a rare and aggressive form of B-cell non-Hodgkin’s lymphoma. Ibrutinib is the third drug approved to treat MCL, following Velcade (2006) and Revlimid (2013), and it is intended for patients with MCL who have received at least one prior therapy. It is the second drug with breakthrough-therapy designation and, therefore, accelerated FDA approval. The FDA also granted ibrutinib priority review and orphan-product designation for MCL because the drug demonstrated the potential to yield significant improvements in safety or effectiveness in the treatment of a serious condition and is intended to treat a rare disease.
The approval of ibrutinib for CLL was based on a multicenter trial of 48 previously treated patients. Ibrutinib 420 mg was administered orally once daily until occurrence of disease progression or unacceptable toxicity. The overall response rate (ORR) and duration of response (DOR) were assessed using a modified version of the International Workshop on CLL criteria by an independent review committee. The ORR was 58.3%, and all responses were partial; none of the patients achieved complete response. The DOR ranged from 5.6 to ≥24.2 months. The accelerated approval of ibrutinib for MCL was based on a study in which 111 participants were administered the drug daily until their disease progressed or adverse effects became intolerable. In this study, nearly 66% of participants experienced cancer shrinkage or disappearance after treatment (ORR). An improvement in survival or disease-related MCL symptoms has not yet been established.
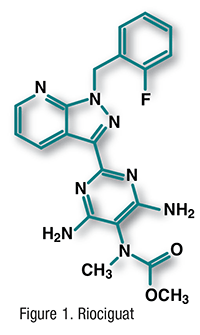
Pharmacology and Pharmacokinetics3,4: Ibrutinib (FIGURE 2) is a pyrazolopyrimidine that functions as a selective inhibitor of Bruton’s tyrosine kinase (BTK). It forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of enzymatic activity and downstream survival pathways activated by this kinase, including extracellular signal-regulated protein kinases 1 and 2, phosphoinositide 3-kinase, and nuclear factor kappa-B. BTK is a signaling molecule of the B-cell antigen receptor and cytokine receptor pathways. BTK’s role in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion. Therefore, ibrutinib inhibition reduces cancer-cell chemotaxis toward the chemokines CXCL12 and -13, inhibits cellular adhesion following stimulation at the B-cell receptor, and drives cells into apoptosis and/or disrupts cell migration and adherence to protective tumor microenvironments. Ibrutinib is the first kinase inhibitor to be approved for CLL.
Ibrutinib is readily absorbed after oral administration, with a median transport maximum of 1 to 2 hours. Administration with food increases ibrutinib exposure approximately twofold. The drug is extensively bound to plasma proteins (97.3%), and its apparent volume of distribution at steady state is approximately 10,000 L. Ibrutinib is metabolized primarily by CYP3A and to a minor extent by CYP2D6, yielding several metabolites. Its dihydrodiol metabolite is pharmacologically active, but less so than the parent drug. The half-life of ibrutinib is 4 to 6 hours, with elimination mainly in the feces (80%). In clinical trials in subjects with hepatic impairment, ibrutinib exposure was approximately sixfold higher in those with moderate hepatic impairment (Child-Pugh class B) compared with mean exposures observed in healthy volunteers.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (>20%) reported in clinical trials were thrombocytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, edema, upper respiratory infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting, and decreased appetite. Other clinically significant adverse reactions included bleeding, infections, kidney problems, and development of other types of cancer. Most adverse reactions were grade 1 or 2, except for cytopenias, infections, and bleeding, which were grade 3. Therefore, patients should be monitored for bleeding events, infection, and fever. Ibrutinib is a substrate of CYP3A; therefore, concurrent administration of strong or moderate CYP3A inhibitors or strong CYP3A inducers should be avoided. If a moderate CYP3A inhibitor must be used, the ibrutinib dosage should be reduced.
Ibrutinib is a Pregnancy Category D drug; therefore, female patients should be apprised of the potential hazard to the fetus. Also, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Dosage and Administration3,4: Ibrutinib is supplied as 140-mg capsules. The recommended dosage for CLL is 420 mg once daily and for MCL is 560 mg once daily. Capsules are to be taken with a glass of water; they should not be chewed or opened. If a moderate CYP3A inhibitor must be used, the dosage should be reduced to 140 mg/day. Ibrutinib should not be used in patients with hepatic impairment.
Obinutuzumab (Gazyva, Genentech)
Indication and Clinical Profile5: The FDA approved obinutuzumab for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL), a blood and bone marrow cancer. According to the National Cancer Institute, 15,680 Americans will be diagnosed with CLL and 4,580 will die from it this year. Obinutuzumab is the first drug with breakthrough-therapy designation to receive FDA approval. The FDA can designate a drug a breakthrough therapy at the request of the sponsor if preliminary clinical evidence indicates that the drug may offer a substantial improvement over available therapies for patients with serious or life-threatening diseases. The FDA also granted obinutuzumab priority review because the drug demonstrated the potential to confer significant improvements in safety or effectiveness in the treatment of a serious condition, and orphan-product designation because the drug is intended to treat a rare disease.
The approval of obinutuzumab for CLL is based on a three-arm, open-label, active-control, randomized multicenter trial comparing obinutuzumab in combination with chlorambucil against chlorambucil alone or rituximab in combination with chlorambucil in 356 participants with previously untreated CLL. The majority of patients received obinutuzumab 1,000 mg on days 1, 8, and 15 of the first cycle, followed by treatment on the first day of 5 subsequent cycles (total of 6 cycles, 28 days each). Chlorambucil 0.5 mg/kg was given orally on days 1 and 15 of all treatment cycles (1-6). Median progression-free survival in the obinutuzumab-plus-chlorambucil arm was 23.0 months versus 11.1 months in the chlorambucil-alone arm. The overall response rate was 75.9% versus 32.1%, complete response was 27.8% versus 0.9%, and median duration of response was 15.2 months versus 3.5 months, respectively, in the obinutuzumab-plus-chlorambucil arm versus the chlorambucil-alone arm.
Pharmacology and Pharmacokinetics5: Obinutuzumab is a monoclonal antibody that targets the CD20 antigen expressed on the surface of pre-B and mature B lymphocytes. When bound to CD20, obinutuzumab mediates B-cell lysis through engagement of immune effector cells, direct activation of intracellular death signaling pathways, and/or activation of the complement cascade. The immune effector cell mechanisms include antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis.
Following IV administration, the mean volume of distribution of obinutuzumab at steady state is approximately 3.8 L. The elimination of obinutuzumab comprises a linear clearance pathway and a time-dependent nonlinear clearance pathway. As treatment progresses, the impact of the time-dependent pathway diminishes in a manner suggesting target-mediated drug disposition. Mean terminal obinutuzumab clearance and half-life are approximately 0.09 L/day (46%) and 28.4 days (43%), respectively. The pharmacokinetics does not appear to be significantly affected by age, weight, or renal function. However, obinutuzumab has not been studied in patients with a baseline creatinine clearance <30 mL/min or in patients with hepatic impairment.
Adverse Reactions and Drug Interactions5: The most common adverse events (>10% incidence) noted in clinical trials of obinutuzumab in combination with chlorambucil were infusion-related reactions, neutropenia, thrombocytopenia, anemia, musculoskeletal pain, and fever. Obinutuzumab carries a boxed warning regarding hepatitis B virus (HBV) reactivation and progressive multifocal leukoencephalopathy, a rare disorder that damages the protective layer of nerves in the white matter of the brain. These are known risks with other monoclonal antibodies in this class, and rare cases were identified in participants in other trials of obinutuzumab. Patients should be advised of these risks, and they should be assessed for HBV and reactivation risk. No formal drug-interaction studies have been conducted with obinutuzumab, which is classified as a Pregnancy Category C drug.
Dosage and Administration5: Obinutuzumab is supplied as a 1,000 mg/40 mL (25 mg/mL) solution in single-use vials. The drug is for IV administration and is given in six cycles of 28 days each. The recommended dosage is 1,000 mg, with the exception of the first infusions in cycle 1, which are administered on day 1 (100 mg) and day 2 (900 mg). Patients should be premedicated with a glucocorticoid, acetaminophen, and an antihistamine to minimize infusion-related reactions. For patients with a high tumor burden and/or high circulating lymphocyte count, tumor lysis syndrome should be anticipated. These patients should be premedicated with antihyperuricemics and adequate hydration. Also, electrolyte abnormalities should be corrected and supportive care provided, including monitoring of renal function and fluid balance. Live vaccinations should not be administered prior to or during therapy with obinutuzumab.
Coagulation Factor XIII A-Subunit (Recombinant) (Tretten, Novo Nordisk)
Indication and Clinical Profile6: Coagulation factor XIII A-subunit (recombinant) (rFXIII-A) was approved as an orphan drug under the FDA’s accelerated approval program for prevention of bleeding in patients with congenital factor XIII A-subunit (FXIII-A) deficiency. FXIII-A deficiency is a rare genetic disorder in which an increased incidence of bleeding results from insufficient synthesis of factor XIII, which is involved in normal clotting. rFXIII-A is the first recombinant product approved for bleeding prophylaxis in adults and children with FXIII-A deficiency.
FDA approval of rFXIII-A was based on a multicenter, open-label, noncontrolled trial in 41 patients aged ≥6 years with known congenital FXIII-A deficiency confirmed by genotyping. All patients received a monthly 35-IU/kg dose of rFXIII-A. Patients had initiated regular replacement therapy ≥6 months before trial entry and had fewer than two treatment-requiring bleeds per year or previously had received only on-demand treatment and had a documented history of at least two treatment-requiring bleeding episodes within the last year. The number of bleeding episodes requiring treatment with an FXIII-containing product was observed, and the mean annual rate of bleeding episodes per subject-year was determined and compared with the historical control group’s estimated bleeding rate. rFXIII-A patients experienced a mean annual rate of bleeding episodes requiring treatment of 0.14 per subject-year, a statistically significant improvement from the historic bleeding rate of 1.68 per subject-year in patients receiving on-demand treatment only. Currently, 34 of these patients and 21 new patients are enrolled in an ongoing second trial that has thus far reported a mean annual rate of bleeding episodes requiring treatment of 0.056 per subject-year.
Pharmacology and Pharmacokinetics6: rFXIII-A is a recombinant form of the catalytic subunit of FXIII, which is the terminal enzyme in the coagulation cascade. FXIII circulates in plasma as a heterotetramer composed of two FXIII A-subunits that function as the catalytic subunit and two FXIII B-subunits that function as carrier molecules. When FXIII is activated by thrombin in the presence of calcium, the A-subunit dissociates from the B-subunit, exposing its active site, which cross-links fibrin and other proteins. This results in a clot with increased mechanical strength and resistance to fibrinolysis, as well as enhanced platelet and clot adhesion.
rFXIII-A has peak plasma concentrations of 0.71 IU/mL and 0.48 IU/mL. It is cleared at a rate of 0.33 mL/h/kg and 0.41 mL/h/kg in adults and children, respectively. It has a respective prolonged half-life of 5.1 days and 7.1 days in adults and children owing to its ability to bind with free B-subunits of FXIII, which protect the drug from inactivation.
Adverse Reactions and Drug Interactions6: The most common adverse reactions (≥1%) reported in clinical trials were headache, pain in extremities, injection-site pain, and elevated fibrin D-dimer levels. No thromboembolic complications were reported during trials; however, such complications may occur, and patients receiving rFXIII-A should be monitored for signs and symptoms of thrombosis, especially if they have conditions that increase the risk of thrombosis formation. The risk of thrombosis is increased if rFXIII-A is administered concomitantly with factor VIIa. During therapy, inhibitory antibodies to rFXIII-A may occur that could result in inadequate response to treatment. An assay measuring FXIII inhibitory antibody concentrations should be performed if breakthrough bleeding occurs or adequate plasma FXIII activity levels are not attained during treatment.
This drug should be given during pregnancy only if it is clearly needed, as it is not known whether rFXIII-A can affect reproductive capacity or causes fetal harm when given during pregnancy (Pregnancy Category C). It should be noted that, during clinical trials, one patient accidentally received 2.3 times the recommended dosage and no clinical signs and symptoms were observed.
Dosage and Administration6: rFXIII-A is supplied as a white lyophilized powder in single-use vials containing 2,000 to 3,125 IU per vial (actual amount stated on each carton and vial), along with a vial of Sterile Water for Injection as a diluent. After reconstitution, each vial contains 667 to 1,042 IU/mL of rFXIII-A to be administered IV at the recommended dosage of 35 IU/kg by a physician or via self-administration. rFXIII-A should be stored away from light and refrigerated at 2°C to 8°C prior to reconstitution. After reconstitution, the solution should be used immediately, but may be stored at 25°C if used within 3 hours. No dosage adjustment is necessary for pediatric patients; however, these patients experienced a greater frequency of adverse reactions. The safety and efficacy of rFXIII-A in elderly patients are not established. rFXIII-A should be discontinued immediately if signs or symptoms of anaphylaxis or hypersensitivity reactions occur.
Collagenase Clostridium Histolyticum (Xiaflex, Auxilium Pharmaceuticals)
Indication and Clinical Profile7: Xiaflex is the first FDA-approved nonsurgical treatment for curvature of the penis (Peyronie disease; PD). In PD, scar tissue develops under the skin of the penis, causing an abnormal bend during erection and resulting in bothersome symptoms, especially during intercourse. Xiaflex was approved in 2010 for the treatment of Dupuytren contracture, a progressive hand disease that can affect a person’s ability to straighten and properly use his or her fingers. Xiaflex is specifically indicated for the treatment of adults with PD with a palpable plaque and curvature deformity ≥30 degrees at therapy initiation.
FDA approval of Xiaflex for PD was based on two controlled trials in 832 adults (studies 1 and 2) with penile curvature deformity of ≥30 degrees in the stable phase of disease. Subjects received up to four treatment cycles of Xiaflex or placebo (weeks 0, 6, 12, 18) and were followed for weeks 24 to 52. In each treatment cycle, two injections of Xiaflex or placebo were administered 1 to 3 days apart, and treatment cycles were repeated at approximately 6-week intervals up to three additional times, for a maximum of eight injection procedures. A penile modeling procedure was performed 1 to 3 days after the second injection of the cycle. The coprimary endpoints were the percentage change from baseline to week 52 in penile curvature deformity and the change from baseline to week 52. Upon completion of study 1, the Xiaflex arm had a –35.0% change, whereas the placebo arm had a –17.8% change. In study 2, the Xiaflex arm had a –33.2% change, whereas the placebo arm had a –21.8% change. Xiaflex also significantly reduced patient-reported bothersome symptoms associated with PD, compared with placebo.
Pharmacology and Pharmacokinetics7: Xiaflex is an injectable formulation of purified collagenase, an enzyme that causes collagen to degrade within the connective tissue. Injection of Xiaflex into a Peyronie collagen plaque appears to result in enzymatic disruption of the plaque. Following disruption of the plaque, penile curvature deformity and bothersome symptoms caused by PD are reduced.
Intralesional administration of Xiaflex results in minimal systemic absorption and distribution. In clinical trials, patients receiving two injections 24 hours apart had only minimal plasma levels, and this effect was short-lived.
Adverse Reactions and Drug Interactions7: The most common adverse reactions associated with the use of Xiaflex for PD were injection-site reactions, including penile hematoma, swelling, and pain. Other less common adverse reactions included lymphadenopathy, itching, edema, and local bruising and bleeding. Allergic reactions were observed in <1% of patients. The drug carries a boxed warning for corporeal rupture (penile fracture) and other serious penile injury, since these events were reported in a small percentage of Xiaflex-treated patients in clinical studies. As a result, for PD treatment, Xiaflex is available only through a restricted Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program requires that participating healthcare professionals and facilities be certified by enrolling and completing training in the administration of Xiaflex for PD. Xiaflex is contraindicated in patients with Peyronie plaques that involve the penile urethra and in those with a history of severe allergic reaction to collagenase used in other therapeutic applications.
No drug-interaction studies have been conducted because the drug does not appear to reach systemic circulation. It is proposed that drugs that interfere with matrix metalloproteinases, such as tetracyclines, anthracyclines, quinolones, and anthraquinone derivatives, could reduce the efficacy of collagenases, but no clinical evidence of such interactions has been reported.
Dosage and Administration7: Xiaflex is supplied in single-use vials containing 0.9 mg of active drug as a sterile powder for reconstitution in a solution for intralesional injection. The recommended treatment involves injection of 0.58 mg Xiaflex into the target plaque once on each of two days, 1 to 3 days apart, according to the injection procedure. For each plaque causing the curvature deformity, up to four treatment cycles may be administered. Treatment cycles may be repeated at approximately 6-week intervals.
REFERENCES
1. Adempas (riociguat) product information. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; May 2014.
2. Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. New Engl J Med. 2013 Jul 25;369:319-329.
3. Imbruvica (ibrutinib) product information. Horsham, PA: Janssen Biotech, Inc; May 2014.
4. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15:48-58.
5. Gazyva (obinutuzumab) product information. South San Francisco, CA: Genentech, Inc; June 2014.
6. Tretten (coagulation factor XIII A-subunit [recombinant]) product information. Plainsboro, NJ: Novo Nordisk Inc; April 2014.
7. Xiaflex (collagenase clostridium histolyticum) product information. Chesterbrook, PA: Auxilium Pharmaceuticals, Inc; December 2013.
To comment on this article, contact rdavidson@uspharmacist.com.



