US Pharm. 2017;42(10):HS35-HS41.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2016–2017 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
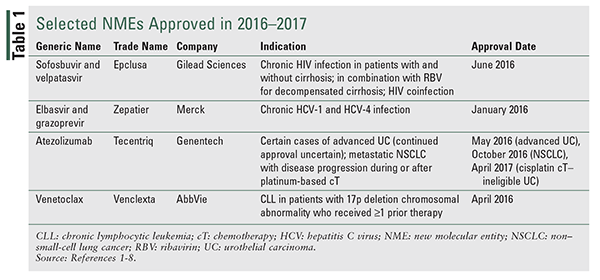
Sofosbuvir and Velpatasvir (Epclusa, Gilead Sciences)
Indication and Clinical Profile1,2: Epclusa is a combination of sofosbuvir—a previously approved hepatitis C virus (HCV) drug—and the new drug velpatasvir. It is indicated as monotherapy to treat chronic HCV infections of genotypes 1 through 6 in patients without cirrhosis or with compensated cirrhosis and as combination therapy with ribavirin (RBV) in patients with decompensated cirrhosis. These indications include patients coinfected with HIV. Approximately 3.2 million Americans are infected with HCV, which causes hepatic inflammation that eventually results in diminished liver function or failure. Often, HCV is asymptomatic for years until liver scarring and cirrhosis develop, causing complications such as bleeding, ascites, infections, jaundice, and liver cancer. Epclusa is the first drug approved by the FDA to treat chronic HCV of all known genotypes, and it is the eighth drug approved to treat chronic HCV.
Approval was based on three randomized phase III trials in 1,558 treatment-naïve and treatment-experienced subjects with HCV (all genotypes represented) who had either no cirrhosis or compensated cirrhosis. The primary endpoint of all trials was sustained virologic response (SVR) at 12 weeks (SVR12; i.e., HCV RNA less than a lower limit of quantification of 25 IU/mL 12 weeks after treatment cessation). In the first trial, which involved subjects with HCV genotype 1, 2, 4, 5, or 6, SVR12 was achieved in 99% of those treated with Epclusa. The second trial, involving subjects with HCV genotype 2, compared SVR12 for Epclusa with SVR at 24 weeks (SVR24) for sofosbuvir plus ribavirin. In this study, Epclusa achieved an SVR12 of 99% versus an SVR24 of 94% for sofosbuvir plus ribavirin. Epclusa’s efficacy in HCV genotype 3 was demonstrated in a third trial, in which 95% of subjects achieved SVR12 versus an SVR24 of 80% in those treated with sofosbuvir plus ribavirin.
Pharmacology and Pharmacokinetics1,2: Velpatasvir (FIGURE 1) is an inhibitor of HCV NS5A protein, which is required for viral replication. Sofosbuvir (FIGURE 1) is a prodrug that is intracellularly converted to an active uridine analogue triphosphate that functions as a chain terminator to viral RNA once it is incorporated into HCV RNA by NS5B polymerase. Both velpatasvir and sofosbuvir have activity across all known HCV genotypes, including strains resistant to protease inhibitors. The actions of velpatasvir and sofosbuvir render HCV unable to replicate.
Velpatasvir achieves its peak plasma concentration at 3 hours and is more than 99.5% plasma protein–bound. It is metabolized via CYP2B6, CYP3A4, and CYP2C8 and does not appear to induce or inhibit metabolism by any major CYP isozyme. Velpatasvir is eliminated primarily in the feces (~94%), with only about 0.4% of the drug eliminated renally. Its half-life is 15 hours.
Sofosbuvir achieves its peak plasma concentration between 0.5 hour and 1 hour and is approximately 60% plasma protein–bound. It is converted to its active metabolite GS-461203 via hydrolysis and phosphoramidate cleavage followed by intracellular phosphorylation by the pyrimidine nucleotide biosynthetic pathway. Sofosbuvir is inactivated via dephosphorylation to its metabolite GS-331007, which cannot be efficiently rephosphorylated. The drug is excreted primarily in the urine (80%), mainly as its GS-331007 metabolite, and has a half-life of 0.5 hours.
Adverse Reactions and Drug Interactions1,2: The most commonly reported adverse effects were headache, fatigue, nausea, asthenia, and insomnia. Patients with decompensated cirrhosis also frequently experienced diarrhea and anemia. Some patients experienced laboratory abnormalities including elevated bilirubin, lipase, and creatine kinase. No adverse effects were noted in animal-reproduction studies; however, because this drug has not been adequately studied in pregnant women, it should be used during pregnancy only if the potential benefit outweighs the potential risk to the fetus. The combination regimen of Epclusa plus RBV is contraindicated in pregnant women and in men whose partner is pregnant.
Velpatasvir’s absorption is pH-dependent; therefore, proton pump inhibitors and antacids should be avoided within 4 hours of Epclusa administration. However, H2-receptor antagonists may be taken simultaneously or 12 hours apart from Epclusa at a dose not exceeding dosages comparable to famotidine 40 mg twice daily. Sofosbuvir and velpatasvir are substrates of P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP) transporters. Velpatasvir is also a substrate of CYP2B6, CYP2C8, and CYP3A4. The use of Epclusa with Pgp inducers and/or moderate-to-potent inducers of CYP2B6, CYP2C8, and CYP3A4 is not recommended, as concurrent use may decrease plasma levels of sofosbuvir and/or velpatasvir, reducing the therapeutic effect of Epclusa. Velpatasvir is an inhibitor of Pgp and BCRP, and it may increase absorption of coadministered substrates of these transporters. Epclusa should not be taken concurrently with amiodarone because of an increased risk of symptomatic bradycardia and fatal cardiac arrest via an unknown mechanism. If Epclusa is coadministered with RBV, all contraindications to ribavirin apply.
Dosage and Administration1,2: Epclusa is supplied as a tablet with a fixed-dose combination of valpatasvir 100 mg and sofosbuvir 400 mg. The recommended dosage is one tablet once daily with or without food. The recommended duration of therapy (including patients coinfected with HIV) is 12 weeks, and patients with decompensated cirrhosis should use Epclusa in combination with RBV. No dose adjustment is recommended in elderly patients or in those with hepatic impairment or creatinine clearance (CrCl) >30 mL/minute/1.73 m2. Epclusa was not studied in pediatric patients, patients with CrCl <30 mL/minute/1.73 m2, or those with end-stage renal disease. Because of the risk of hepatic virus reactivation, all patients should be tested for evidence of current or prior HBV infection before HCV treatment is initiated.
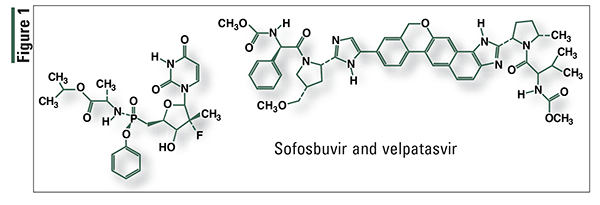
Elbasvir and Grazoprevir (Zepatier, Merck)
Indication and Clinical Profile3,4: Zepatier is a combination of two newly approved hepatitis C virus (HCV) drugs: elbasvir and grazoprevir. It is indicated to treat chronic HCV genotype 1 (HCV-1) and HCV genotype 4 (HCV-4) infection. HCV infection was described briefly in the previous section. Approximately 71 million people globally have chronic HCV; 95% of cases are estimated to be curable with available medications, but access to diagnosis and treatment is low. Zepatier is the seventh drug approved by the FDA to treat chronic HCV since the approval of simeprevir (Olysio) in November 2013.
Approval of Zepatier was based on six randomized studies in 1,401 subjects with HCV-1 or HCV-4 infection, including patients who had failed prior therapy with peginterferon alfa and ribavirin (RBV) with or without an HCV NS3/4A protease inhibitor and patients who were treatment naïve, as well as those with compensated cirrhosis or chronic kidney disease and those on dialysis. The primary endpoint of all trials was sustained virologic response (SVR) after 12 or 16 weeks of treatment (SVR12 and SVR16), defined as HCV RNA levels less than the lower limit of quantification. Zepatier achieved high rates of SVR across all studies, with a range of 92% to 95%, 96% to 100%, and 97% to 100% of patients attaining SVR using the preferred regimen and duration of therapy based on patient characteristics for HCV-1a, HCV-1b, and HCV-4 infections, respectively.
Pharmacology and Pharmacokinetics3,4: Elbasvir (FIGURE 2) is an inhibitor of HCV NS5A protein, which is required for viral replication. Grazoprevir (FIGURE 2) is an inhibitor of HCV NS3/4A protease, which is necessary for proteolytic cleavage of HCV-encoded polyproteins used for viral replication. The actions of both elbasvir and grazoprevir interrupt the replication of HCV.
Elbasvir is 32% bioavailable, achieves peak plasma concentration between 3 and 6 hours, and is 99.8% plasma protein bound. Grazoprevir is 27% bioavailable, achieves peak plasma concentration between 0.5 and 3 hours, and is about 99.9% plasma protein bound. Both elbasvir and grazoprevir are partially metabolized via oxidation by CYP3A4, with grazoprevir acting as a weak inhibitor of this enzyme. Both drugs are eliminated primarily in the feces (>90%), with <1% of the dose recovered in urine. The half-lives of elbasvir and grazoprevir are 24 and 31 hours, respectively.
Adverse Reactions and Drug Interactions3,4: The most common adverse effects reported in clinical trials were headache, fatigue, and nausea. Some patients also experienced laboratory abnormalities including elevated bilirubin, alanine aminotransferase, and hemoglobin. No adverse effects were noted in animal-reproduction studies of Zepatier; however, because this drug has not been adequately studied in pregnant women, it should be used during pregnancy only if the potential benefit outweighs the potential risk to the fetus. If Zepatier is administered with RBV, the combination regimen is contraindicated in pregnant women and in men whose partner is pregnant.
Both elbasvir and grazoprevir are substrates of CYP3A. Coadministration with strong CYP3A inducers and efavirenz, a moderate CYP3A inducer, is contraindicated because metabolic induction can decrease serum concentrations of both components, resulting in loss of virologic response. The grazoprevir component is a substrate of organic anion-transporting polypeptides 1B1/3 (OATP1B1/3) transporters, so concurrent use with OATP1B1/3 inhibitors that are known or expected to significantly increase grazoprevir plasma concentrations is contraindicated. If Zepatier is coadministered with RBV, all contraindications to RBV apply.
Dosage and Administration3,4: Zepatier is supplied as a tablet of a fixed-dose combination of elbasvir 50 mg and grazoprevir 100 mg. The recommended dosage is one tablet once daily with or without food. No dose adjustment is recommended in elderly patients or in those with renal impairment or mild (Child-Pugh grade A) hepatic impairment. Zepatier is contraindicated in patients with moderate-to-severe hepatic impairment (Child-Pugh grade B-C). Patients with HCV-1a should be tested for NS5A resistance–associated polymorphisms prior to treatment initiation to determine the regimen and duration. Treatment-naïve patients with HCV-4, HCV-1b, and HCV-1a without baseline NS5A polymorphisms should receive 12 weeks of therapy. Patients with HCV-1 without baseline NS5A polymorphisms who have failed treatment with peginterferon plus RBV plus an HCV NS3/4A protease inhibitor should receive 12 weeks of therapy in combination with RBV. HCV-4 patients who have been treated with peginterferon plus RBV, as well as those with HCV-1a with baseline NS5A polymorphisms (regardless of treatment experience), should receive 16 weeks of therapy in combination with RBV. Because of the risk of hepatic virus reactivation, all patients should be tested for evidence of current or prior HBV infection before initiation of HCV treatment.
Atezolizumab (Tecentriq, Genentech)
Indication and Clinical Profile5,6: In October 2016, atezolizumab was approved for the treatment of metastatic non–small cell lung cancer (NSCLC) patients with disease progression during or following platinum-containing chemotherapy who have progressed on an appropriate FDA-approved targeted therapy if their tumor had epidermal growth factor receptor or anaplastic lymphoma kinase gene abnormalities. Previously (May 2016), this drug had received accelerated approval for treatment of locally advanced or metastatic urothelial carcinoma (mUC) in patients with disease progression during or following any platinum-containing chemotherapy or within 12 months of receiving chemotherapy before surgery (neoadjuvant) or after surgery (adjuvant). In April 2017, the drug’s indications were extended to include treatment of locally advanced UC or mUC in patients ineligible for cisplatin chemotherapy. However, at this time, continued approval of atezolizumab for advanced UC or mUC is uncertain based on recent disappointing results in phase III confirmatory trials. Bladder cancer is the most common type of UC (other types include cancers of the urethra, ureters, and renal pelvis). Up to one-half of all people with advanced bladder cancer are unable to receive cisplatin chemotherapy as initial treatment and therefore have a high unmet medical need.
The approval of atezolizumab for metastatic NSCLC was based on results of the phase III OAK and phase II POPLAR studies. OAK was a global, multicenter, controlled study of 1,225 subjects who were randomized 1:1 to receive either docetaxel (75 mg/m2 IV infusion) or atezolizumab (1,200 mg IV infusion) every 3 weeks. In this study, subjects in the overall study population lived a median of 13.8 months, 4.2 months longer than those who received docetaxel chemotherapy (median overall survival, 13.8 vs. 9.6 months). OAK enrolled subjects regardless of their programmed death-ligand 1 (PD-L1) status and included both squamous and nonsquamous disease types. In POPLAR, a randomized study of 287 patients, atezolizumab doubled the likelihood of overall survival in subjects whose cancer expressed the highest levels of PD-L1, compared with subjects receiving docetaxel chemotherapy. Improved survival was also observed in subjects who had medium, high, or any level of PD-L1 expression.
The approval of atezolizumab for mUC was based on a single-arm clinical trial involving 310 patients with locally advanced or metastatic disease. This trial measured the percentage of patients who experienced complete or partial tumor shrinkage (objective response rate). The study also examined the difference in effect, based on positive versus negative expression of PD-L1 protein, on patients’ tumor-infiltrating immune cells. In all patients, 14.8% experienced at least partial tumor shrinkage, an effect that lasted from more than 2.1 months to more than 13.8 months at the time of response analysis. In patients who were classified as positive for PD-L1 expression, 26% experienced a tumor response, compared with 9.5% of patients who were classified as negative for PD-L1 expression.
Pharmacology and Pharmacokinetics5,6: Atezolizumab is a PD-L1–blocking antibody. PD-L1 is expressed on some tumor cells or tumor-infiltrating immune cells and may contribute to inhibition of the antitumor immune response in the tumor microenvironment. Binding of PD-L1 to the PD-1 and B7.1 receptors found on T cells and antigen-presenting cells suppresses cytotoxic T-cell activity, T-cell proliferation, and cytokine production. Atezolizumab’s blocking of PD-L1 to its receptors therefore releases PD-L1/PD-1-mediated inhibition of the immune response, including activation of the antitumor immune response, without inducing antibody-dependent cellular cytotoxicity. For some cancers, notably bladder cancer, the probability of benefit is related to PD-L1 expression, but many cancers with PD-L1 expression do not respond, and about 15% of those without PD-L1 expression do respond.
Exposure to atezolizumab increases the dosage proportionally over the range of 1 mg/kg to 20 mg/kg, including the fixed-dose 1,200 mg administered every 3 weeks. Based on a population analysis, the typical clearance is 0.20 L/day, the volume of distribution at steady state is 6.9 L, and the terminal half-life is 27 days. Steady state is obtained after 6 to 9 weeks (2-3 cycles) of repeated dosing. The systemic accumulation in AUC, maximum concentration, and trough concentration are 1.91-fold, 1.46-fold, and 2.75-fold, respectively. In a post hoc analysis, atezolizumab clearance was found to decrease over time, with a mean maximal reduction from baseline value of approximately 17%; however, the decrease in clearance is not considered clinically relevant. Age, weight, sex, positive antitherapeutic antibody status, albumin levels, tumor burden, race, mild-to-moderate renal impairment (estimated glomerular filtration rate [GFR] 30-89 mL/min/1.73 m2), mild hepatic impairment, level of PD-L1 expression, and Eastern Cooperative Oncology Group status do not appear to have a clinically significant effect on the systemic exposure of atezolizumab. The effect of severe renal impairment (estimated GFR 15-29 mL/minute/1.73 m2) or moderate-to-severe hepatic impairment on the pharmacokinetics of atezolizumab is unknown.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions (20% or more) attributed to atezolizumab in clinical trials were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. The most common (2% or more) grade 3 or 4 adverse events in atezolizumab-treated patients were dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, anemia, musculoskeletal pain, aspartate aminotransferase increase, alanine aminotransferase increase, dysphagia, and arthralgia. Clinically significant immune-related adverse events in patients receiving atezolizumab included pneumonitis, hepatitis, colitis, and thyroid disease. To date, no drug interactions with atezolizumab have been documented.
Dosage and Administration5,6: Atezolizumab is a sterile, preservative-free, and colorless to slightly yellow solution for IV infusion supplied as a carton containing one 1,200 mg/20 mL single-dose vial. Atezolizumab should not be administered as an IV push or bolus. The recommended dosage is 1,200 mg administered as an IV infusion over 60 minutes every 3 weeks until disease progression or unacceptable toxicity. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes. Generally, subsequent doses of atezolizumab should be withheld if drug-related grade 2 or higher toxicities develop. The prescribing information contains more detailed information concerning withholding or stopping atezolizumab therapy when specific adverse effects emerge.
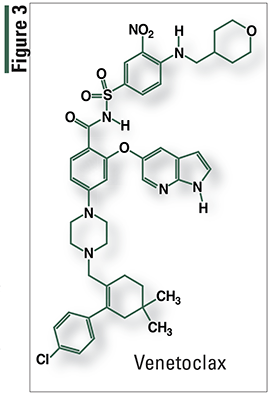
Venetoclax (Venclexta, AbbVie)
Indication and Clinical Profile7,8: Venetoclax was approved for the treatment of chronic lymphocytic leukemia (CLL) patients with a 17p deletion chromosomal abnormality who have undergone at least one prior therapy. It is the first FDA-approved treatment that targets the B-cell lymphoma 2 (BCL-2) protein, which supports cancer-cell growth and is overexpressed in many patients with CLL. According to the National Cancer Institute, CLL is one of the most common types of leukemia in adults; approximately 15,000 new cases are diagnosed each year. CLL patients who have a 17p deletion lack a portion of the chromosome that acts to suppress cancer growth. This chromosomal abnormality occurs in approximately 10% of patients with untreated CLL and in approximately 20% of those with relapsed CLL. Venetoclax is indicated after detection of a 17p deletion is confirmed through the use of the companion diagnostic Vysis CLL FISH Probe Kit. In the past few years, two other drugs—Imbruvica (ibrutinib) and Zydelig (idelalisib)—also have been approved to treat CLL.
The efficacy of venetoclax was demonstrated in a single-arm clinical trial of 106 CLL patients with a 17p deletion who had undergone at least one prior therapy. Patients received oral venetoclax daily, starting at 20 mg and increasing over a 5-week period to 400 mg; they continued to receive venetoclax 400 mg daily until disease progression or unacceptable toxicity occurred. The efficacy of venetoclax was evaluated by overall response rate (ORR) as assessed by an independent review committee using the International Workshop for Chronic Lymphocytic Leukemia update of National Cancer Institute Working Group–sponsored guidelines. The ORR was 85% and the median time to first response was 0.8 months. Response duration ranged from 2.9 to 19.0 months or more. Based on these results, the FDA granted venetoclax breakthrough therapy designation, priority review status, and accelerated approval for CLL. (These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.) Venetoclax also received orphan drug designation.
Pharmacology and Pharmacokinetics7,8: Venetoclax (FIGURE 3) is a selective and orally bioavailable small-molecule inhibitor of BCL-2, an antiapoptotic protein. BCL-2 overexpression has been demonstrated in CLL cells, where it mediates tumor-cell survival and has been associated with resistance to chemotherapeutic agents. Venetoclax helps restore apoptosis by binding directly to the BCL-2 protein, displacing proapoptotic proteins like BIM and triggering mitochondrial outer-membrane permeabilization and activation of caspases.
Upon oral administration, maximum plasma concentrations of venetoclax are achieved within 5 to 8 hours. The steady-state maximum concentration with low-fat meals at a dosage of 400 mg once daily is about 2.1 mcg/mL. Venetoclax is highly bound to human plasma proteins, and its apparent volume of distribution exceeds 200 L. Venetoclax is metabolized by CYP3A4/5; therefore, patients taking this drug should not consume grapefruit products because they contain CYP3A inhibitors. Additionally, the concurrent use of other CYP3A inhibitors (i.e., erythromycin, ciprofloxacin, diltiazem, dronedarone, fluconazole, verapamil) should be avoided. Venetoclax is excreted primarily via the fecal route. No specific clinical trials have been conducted in subjects with hepatic impairment, but as noted above, venetoclax is eliminated via the liver. Presently, no dose adjustment is recommended in patients with mild-to-moderate hepatic impairment, but population pharmacokinetic analyses suggest the potential for increased adverse events in patients with moderate hepatic impairment.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions associated with venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, fatigue, and fertility problems (males). Serious complications include pneumonia, neutropenia, fever, autoimmune hemolytic anemia, anemia, and metabolic abnormalities known as tumor lysis syndrome (TLS), which is associated with rapid tumor reduction. Because of the risk of neutropenia, blood counts and signs of infection should be monitored and live attenuated vaccines should be avoided. To minimize the risk of TLS, especially during the initial dosing ramp-up phase (5 weeks), patients should be premedicated with antihyperuricemics and adequate hydration should be ensured; this may require IV hydration in patients at greater risk for TLS. Venetoclax therapy may have to be interrupted in patients experiencing grade 3 or 4 neutropenia with infection or fever, grade 4 hematologic toxicities (except lymphopenia), or other grade 3 or 4 nonhematologic toxicities. Once the toxicity has resolved to grade 1 or baseline level, venetoclax therapy may be resumed. No data are available on the use of venetoclax in pregnant women, but this drug has been shown to cause embryo-fetal harm in animal models; therefore, females of reproductive potential should be advised of the potential fetal risk and to use effective contraception during treatment.
Venetoclax is a CYP 3A4/5 substrate, and concomitant use with strong inhibitors of CYP3A at initiation and during the ramp-up phase is contraindicated. Also, the concurrent use of venetoclax with moderate CYP3A inhibitors, strong or moderate CYP3A inducers, P-glycoprotein (Pgp) inhibitors, or narrow-therapeutic-index Pgp substrates should be avoided. If a moderate CYP3A inhibitor or a Pgp inhibitor must be used, the dose of venetoclax should be reduced by at least 50%. If concurrent therapy with a strong CYP3A inhibitor is required after the ramp-up phase, the venetoclax dose should be reduced by at least 75%. If a narrow-therapeutic-index Pgp substrate must be used, it should be taken at least 6 hours before venetoclax. Patients should avoid consuming grapefruit, Seville oranges, and starfruit, as these foods are known to inhibit CYP3A4 metabolism.
Dosage and Administration7,8: Venetoclax is supplied as 10-, 50-, and 100-mg tablets for oral administration. Therapy should be initiated at 20 mg once daily for 7 days, followed by a weekly ramp-up dosing schedule to the recommended daily dose of 400 mg. The tablets should be taken orally once daily with a meal and water; they should not be chewed, crushed, or broken. Also, prophylactic measures as described above should be instituted to reduce the risk of TLS.
REFERENCES
1. Epclusa (sofosbuvir and velpatasvir) package insert. Foster City, CA: Gilead Sciences; August 2017.
2. Feld JJ, Jacobson IM, Hézode C, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. New Engl J Med. 2015;373:2599-2607.
3. Zepatier (elbasvir and grazoprevir) package insert. Whitehouse Station, NJ: Merck and Co, Inc; February 2017.
4. Bell AM, Wagner JL, Barber KE, Stover KR. Elbavir/grazoprevir: a review of the latest agent in the fight against hepatitis C. Int J Hepatol. 2016;2016:3852126.
5. Tecentriq (atezolizumab) package insert. South San Francisco, CA: Genentech, Inc; April 2017.
6. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909-1920.
7. Venclexta (venetoclax) package insert. North Chicago, IL: AbbVie Inc; April 2016.
8. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768-778.
To comment on this article, contact rdavidson@uspharmacist.com.



