US Pharm. 2023;48(10):HS11-HS16.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2022–2023 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included are brief summaries of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of an NME’s therapeutic profile are not detected in premarketing clinical studies and emerge after the drug is used in the broader population. For example, previously unreported adverse reactions become apparent for some NMEs within several years of their marketing. Some NMEs may eventually acquire at least one black box warning for serious drug reactions or are withdrawn from the market for safety reasons not recognized at the time of approval. Therefore, while this review offers an introduction to some new drugs, it is essential that practitioners be aware of changes in their therapeutic profiles as reported in the pharmaceutical literature and by patients.
Daprodustat (Jesduvroq, GlaxoSmithKline)
Indication and Clinical Profile1,2: Daprodustat, a hypoxia-inducible factor–prolyl hydroxylase (HIF-PH) inhibitor, is indicated for treatment of anemia due to chronic kidney disease (CKD) in adults who have been on dialysis for >4 months. The development of anemia (hemoglobin [Hb] <13.0 g/dL for men, <12.0 g/dL for nonpregnant women) is common in CKD patients, and standard-of-care treatment includes IV iron, erythropoiesis-stimulating agents (ESAs), and RBC transfusions. A developing potential treatment target is inhibition of HIF, which is responsible for regulation of genes involved in erythropoiesis and iron metabolism, promotion of iron absorption, iron transport, and heme synthesis. Under normoxic conditions, HIF-PH inhibitors increase HIF degradation.
The efficacy and safety of daprodustat were evaluated in ASCEND-D, a randomized, sponsor-blind, active-controlled, global, multicenter, event-driven clinical trial. Subjects had been on dialysis for >4 months and were stratified by dialysis type. With 1:1 randomization, the hemodialysis (HD) group received either oral daprodustat or IV epoetin alfa and the peritoneal dialysis (PD) group received either either oral daprodustat or SC darbepoetin alfa. The coprimary efficacy and safety endpoints were mean change in Hb from baseline to the evaluation period (weeks 28-52) and time to first adjudicated MACE (all-cause mortality, nonfatal infarction, or nonfatal stroke). Noninferiority of daprodustat to epoetin alfa and darbepoetin alfa was achieved if the lower limit of the 95% CI for the overall Hb treatment difference was greater than the prespecified noninferiority margin of –0.75 g/dL; for time to first adjudicated MACE, noninferiority of daprodustat was achieved if the upper limit of the 95% CI was less than the prespecified noninferiority margin of 1.25.
Each treatment arm followed a protocol-specified dose-adjustment algorithm to achieve and/or maintain an Hb target of 10 g/dL to 11 g/dL. Daprodustat dosing was initiated at 4 mg, 6 mg, 8 mg, or 12 mg orally once daily, based on the prior ESA dose. Initial dosing for epoetin alfa (HD patients) or darbepoetin alfa (PD patients) was the patient’s current dose rounded to the nearest study dose; patients receiving another ESA were switched to the nearest equivalent epoetin alfa or darbepoetin alfa dose. Median dosages at week 52 were 6 mg/day for daprodustat, 8,000 units/week for epoetin alfa, and 150 mcg every 4 weeks for darbepoetin alfa. The overall mean change in Hb was 0.3 g/dL in the daprodustat group versus 0.1 g/dL in the group comprising epoetin alfa and darbepoetin alfa patients (95% CI, mean treatment difference, 0.2 [0.1, 0.2]). The second endpoint for MACE showed a hazard ratio of 0.93.
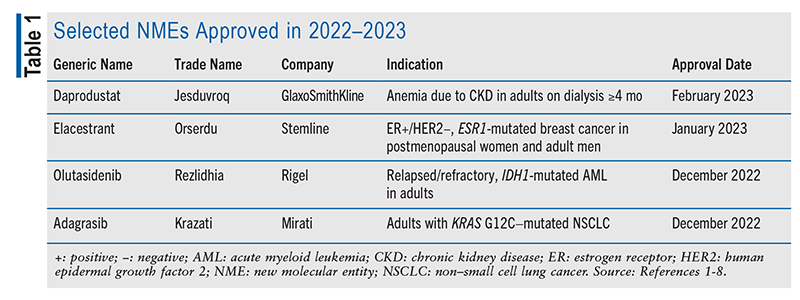
Pharmacology and Pharmacokinetics1,2: Daprodustat (FIGURE 1) is a trioxopyrimdine-glycine compound that increases the production of erythropoietin through reversible inhibition of HIF-PH1, -PH2, and -PH3. This effect is dose-dependent and occurs 6 to 8 hours after administration. Continuous use results in increased reticulocyte count between 7 and 15 days and in new Hb steady-state levels after several weeks.
Daprodustat is readily absorbed, with a time to peak concentration of 1 to 4 hours, and steady-state concentrations are achieved within 24 hours. Calorie and fat intake do not appear to effect absorption. Daprodustat has an approximately equal distribution between plasma and blood cells (blood:plasma ratio, 1.23). Steady-state volume of distribution post IV administration is 14.3 L, and plasma protein binding is >99%. The terminal half-life is 1 to 4 hours. Metabolism is cytochrome-based, with 95% occurring via CYP2C8 and 5% via CYP3A4. Daprodustat is excreted primarily (99.5%) as oxidative metabolites, with 74% in the feces and 21% in the urine.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions reported for daprodustat (>10% of patients) included exacerbation of hypertension, thrombotic vascular events, and abdominal pain. Warnings and precautions include risk of hospitalization for heart failure, hypertension, gastrointestinal erosion, serious adverse events in patients with anemia due to CKD who are not on dialysis, and malignancy. This agent carries a black box warning for increased risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access.
Daprodustat is primarily metabolized by CYP2C8, and it is significantly altered by CYP2C8 inducers and inhibitors. Daprodustat is contraindicated in patients taking strong CYP2C8 inhibitors. The starting dose of daprodustat must be reduced by half when it is being initiated in patients taking clopidogrel or a moderate CYP2C8 inhibitor. Patients taking CYP2C8 inducers should be monitored because of the potential for loss of daprodustat efficacy.
Dosage and Administration1: Daprodustat is supplied as tablets of 1 mg, 2 mg, 4 mg, 6 mg, and 8 mg for oral administration. Prior to initiation of daprodustat, an evaluation of iron status and liver testing is recommended. For patients not receiving ESA treatment prior to initiation, daprodustat initiation is based on the Hb level, with a dose of 4 mg recommended for Hb <9 g/dL, 2 mg for Hb>9 g/dL to <10 g/dL, and 1 mg for Hb >10 g/dL. For patients who are already receiving treatment with an ESA, switching from an ESA to daprodustat is based on the dose regimen of the ESA per the recommendations of the manufacturer.
Elacestrant (Orserdu, Stemline)
Clinical Indication and Clinical Profile3,4: Elacestrant is an estrogen antagonist indicated for treatment of estrogen receptor–positive, human epidermal factor growth receptor 2–negative (ER+/HER2–), ESR1-mutated breast cancer. Elacestrant is FDA approved for use in postmenopausal women and adult men with ER+/HER2–, ESR1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy.
The efficacy and safety of elacestrant were evaluated in EMERALD, a randomized, multicenter trial in 478 patients (postmenopausal women and adult men) with ER+/HER– advanced or metastatic breast cancer, 228 of whom had ESR1 mutations. Patients received either elacestrant 345 mg orally once daily (n = 239) or investigator’s choice of endocrine therapy (fulvestrant or aromatase inhibitor; n = 239), with therapy continuing until disease progression or unacceptable toxicity. The efficacy outcomes included progression-free survival (PFS) and overall survival. PFS was assessed by a blinded imaging-review committee. The number of PFS events was 62 in the elacestrant group and 78 in the endocrine therapy group (hazard ratio 0.55).
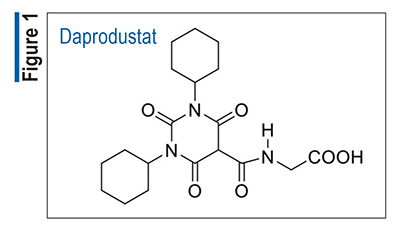
Pharmacology and Pharmacokinetics3,4: Elacestrant (FIGURE 2) is a tetrahydronaphthalene-methoxybenzene compound that functions as an estrogen receptor-alpha antagonist. In ER+/HER2– breast cancer cells, elacestrant inhibited 17-beta-estradiol mediated cell proliferation at concentrations inducing degradation of estrogen receptor-alpha protein mediated through the proteasomal pathway. In vitro and in vivo antitumor activity, including ER+/HER2– breast cancer models resistant to fulvestrant and cyclin-dependent kinase 4/6 inhibitors and those harboring ESR1 mutations, was observed.
When administered at the recommended dosage, elacestrant exhibits a maximum concentration of drug (Cmax) of 119 ng/mL and an AUC0-24h of 2,440 ng * h/mL. Time to achieve peak plasma concentration ranges from 1 to 4 hours. Bioavailability is 10%, and administration with a high-fat meal increases Cmax and AUC by 42% and 22%, respectively. The estimated apparent volume of distribution is 5,800 L, and plasma protein binding is >99%. The elimination half-life is 30 to 50 hours, with an estimated mean clearance of 186 L/hour and renal clearance of <0.14 L/hour. Elacestrant is metabolized primarily by CYP3A4. Most of the drug and metabolites are eliminated in the feces (82%); 7.5% is eliminated in the urine.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions that were reported for elacestrant (>10% of patients) were abdominal pain, constipation, decreased appetite, diarrhea, nausea, vomiting, fatigue, headache, musculoskeletal pain, and hot flashes. Commonly reported laboratory abnormalities (>10% of patients) included decreased serum sodium, hypercholesterolemia, hypertriglyceridemia, decreased hemoglobin, and increased serum creatinine.
There are no contraindications for elacestrant; however, warnings and precautions include dyslipidemia and embryo-fetal toxicity. Concomitant use with strong or moderate CYP3A4 inducers and inhibitors should be avoided. Elacestrant inhibits P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP); therefore, dose reductions in Pgp and BCRP substrates are recommended. Elacestrant may cause fetal harm when administered to a pregnant woman. Verification of pregnancy status prior to initiation is recommended, and female patients should be counseled on the use of effective contraception during treatment and for 1 week after the last dose. Elacestrant should not be used while breastfeeding or in patients with severe hepatic impairment (Child-Pugh class C). Dose reductions are recommended in patients with moderate hepatic impairment (Child-Pugh class B).
Dosage and Administration3,4: Elacestrant is supplied as tablets of 345 mg (equivalent to 400 mg elacestrant dihydrochloride) and 86 mg (equivalent to 100 mg elacestrant dihydrochloride). The recommended dosage is 345 mg orally with food once daily until disease progression or unacceptable toxicity occurs. Tablets should be taken whole and not chewed, crushed, or split. It is recommended to take elacestrant with food to reduce nausea and vomiting. If a dose is vomited, the next dose should be taken at the next scheduled time. In the event of adverse reactions, a first-dose reduction to 258 mg once daily is recommended, followed by a second-dose reduction to 172 mg once daily in the event of continued adverse reactions. Elacestrant should be permanently discontinued if further dose deescalation is required.
Olutasidenib (Rezlidhia, Rigel)
Indication and Clinical Profile5,6: Olutasidenib, a small-molecule inhibitor of mutated isocitrate dehydrogenase-1 (IDH1), is indicated for treatment of adults with relapsed or refractory acute myeloid leukemia (AML) with a susceptible IDH1 mutation. IDH genes are responsible for converting isocitrate to alpha-ketoglutarate (AKG). IDH mutation leads to the production of an abnormal metabolite, 2-hydroxyglutarate (2HG), which results in leukemogenesis. IDH1 mutations occur in 6% to 10% of all AML cases, and drugs that inhibit production of hydroxyglutaric acid may reduce the effects of AKG-dependent enzymes.
Olutasidenib’s efficacy and safety were assessed in an open-label, single-arm, multicenter clinical trial of 147 adults with relapsed or refractory AML with an IDH1 mutation. Patients received olutasidenib 150 mg orally twice daily until disease progression, development of unacceptable toxicity, or hematopoietic stem-cell transplantation. Efficacy outcomes were rate of complete remission (CR) plus CR with partial hematologic recovery (CR + CRh), duration of CR + CRh, and rate of conversion from transfusion dependence to transfusion independence. For the efficacy endpoints, CR + CRh was 51, with a median duration of response (DOR) of 25.9 months; CR was 47, with a median DOR of 28.1 months; and CRh was 4.
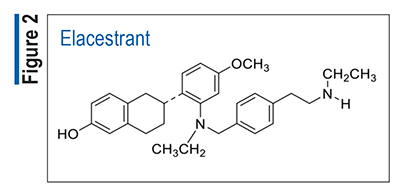
Pharmacology and Pharmacokinetics5,6: Olutasidenib (FIGURE 3) is a dihydroquinoline-dihydropyridine derivative that inhibits IDH1. IDH1 mutations may be responsible for increased levels of 2HG in leukemia cells, where it promotes neoplastic cell proliferation. The inhibition of IDH1 decreases 2HG production, and it helps restore normal cell differentiation.
At recommended doses, median time to maximum concentration is 4 hours. Administration with a high-fat meal increases the maximum concentration by 191% and AUC exposure by 83%. The mean volume of distribution is 319 L, and plasma protein binding is 93%. Metabolism occurs primarily by CYP3A4. The mean plasma half-life is 67 hours, and oral clearance is 4 L/hour. No clinically significant differences in pharmacokinetics based on age, sex, body weight, or mild-to-moderate renal or hepatic impairment were observed. The effect of severe renal impairment or hepatic impairment on pharmacokinetics has not been determined.
Adverse Reactions and Drug Interactions5,6: Common adverse reactions reported for olutasidenib (≥20% of patients) included skin rash, constipation, diarrhea, nausea, stomatitis, fatigue, malaise, dyspnea, and fever. Commonly reported laboratory abnormalities (>20% of patients) included decreased serum potassium, decreased serum sodium, increased uric acid, increased serum alanine aminotransferase, increased serum alkaline phosphatase, increased serum aspartate aminotransferase, increased serum bilirubin, and increased serum creatinine.
Warnings and precautions for olutasidenib include differentiation syndrome (black box warning) and hepatotoxicity. Olutasidenib should not be used in pregnant women, based on its potential to cause fetal harm. The drug is excreted in breast milk, and breastfeeding should be avoided during therapy and for 2 weeks after the last dose. Because olutasidenib is a CYP3A substrate, the concurrent administration of CYP3A inducers should be avoided. Olutasidenib is also a CYP3A inducer, and therefore it should not be used concurrently with sensitive CYP3A substrates because this may reduce their efficacy.
Dosage and Administration5,6: Olutasidenib is supplied as 150-mg tablets. The recommended dosage is 150 mg orally twice daily until disease progression or unacceptable toxicity. Patients without disease progression should be treated for ≥6 months to allow time for clinical response. Olutasidenib should be taken on an empty stomach at the same time each day.
Adagrasib (Krazati, Mirati)
Indication and Clinical Profile7,8: Adagrasib is a KRAS G12C inhibitor indicated for treatment of KRAS G12C–mutated locally advanced or metastatic non–small cell lung cancer (NSCLC). One gene that commonly is mutated in pancreatic, lung, and colorectal cancer is KRAS. KRAS G12C is a single-point mutation that has a glycine-to-cysteine substitution at codon 12. Treatment directed at KRAS G12C is a potential targeted drug therapy for patients with this mutation. Adagrasib was granted accelerated approval for use in adults with KRAS G12C–mutated locally advanced or metastatic NSCLC—as determined by an FDA-approved test—who have received at least one prior systemic therapy.
The efficacy of adagrasib was evaluated in KRYSTAL-1, a multicenter, single-arm, open-label, expansion cohort study of 112 patients with KRAS G12C–mutated NSCLC. Enrolled patients had previously received treatment with a platinum-based regimen and an immune checkpoint inhibitor, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and had at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1. Patients received adagrasib 600 mg orally twice daily until acceptable toxicity or disease progression. The primary efficacy outcomes included confirmed objective response rate (ORR) and duration of response (DOR). Outcomes were evaluated by blinded independent central review. In the study population, the ORR was 43% (95% CI, 34-53) and the DOR was a median of 8.5 months (95% CI, 6.2-13.8).
Pharmacology and Pharmacokinetics7: Adagrasib (FIGURE 4), a highly functionalized tetrahydropyrido-pyrimidine, is an irreversible inhibitor of KRAS G12C. Adagrasib covalently binds to the mutant cysteine in KRAS G12C, keeping the mutant KRAS protein in its inactive state while not affecting wild-type KRAS protein. This results in inhibited tumor-cell growth with resulting tumor regression in KRAS G12C–mutated tumor xenograft models, with minimal off-target activity.
For the dose range of 400 mg to 600 mg, adagrasib has dose-dependent effects on AUC and maximum concentration of drug. Steady state is reached within 8 days, and accumulation is sixfold. Median time to maximum drug concentration is 6 hours, and the volume of distribution is 942 L (57%). Human plasma protein binding is 98% in vitro. Adagrasib is metabolized primarily by CYP3A4, and 75% is excreted in the feces and 4.5% in the urine. The terminal elimination half-life is 23 hours.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions reported for adagrasib (>20% of patients) were edema, abdominal pain, constipation, decreased appetite, diarrhea, vomiting, dizziness, fatigue, musculoskeletal pain, hepatotoxicity, renal insufficiency, cough, dyspnea, pneumonia, and prolonged QT interval. Common laboratory abnormalities (>25% of patients) included decreased serum albumin, decreased serum magnesium, decreased serum potassium, increased serum lipase, decreased platelet count, lymphocytopenia, increased serum alanine aminotransferase, increased serum aspartate aminotransferase, and increased serum creatine phosphokinase.
There are no contraindications; however, warnings and precautions include gastrointestinal toxicity, hepatotoxicity, pulmonary toxicity, and QTc-interval prolongation. No data are available on adagrasib use in pregnant women. Because it is a CYP3A4 substrate and inhibitor, adagrasib should be avoided in patients taking CYP3A4 inducers. Adagrasib also inhibits CYP2C9, CYP2D6, and P-glycoprotein; therefore, concurrent use of sensitive substrates should be avoided to reduce the risk of drug interactions.
Dosage and Administration7: Adagrasib is supplied as tablets of 200 mg. The recommended dosage is 600 mg orally twice daily until disease progression or unacceptable toxicity. Adagrasib should be taken at the same time each day, with or without food, and the tablet should be swallowed whole. In the event of adverse reactions, the dosage of adagrasib may be reduced to 400 mg twice daily, then reduced to 600 mg once daily. Adagrasib should be discontinued in patients unable to tolerate 600 mg once daily.
REFERENCES
1. Jesduvroq (daprodustat) product information. Durham, NC: GlaxoSmithKline; August 2023.
2. Hanna RM, Streja E, Kalantar-Zadeh K. Burden of anemia in chronic kidney disease: beyond erythropoietin. Adv Ther. 2021;38(1):52-75.
3. Orserdu (elacestrant) product information. New York, NY: Stemline Therapeutics, Inc; January 2023.
4. Elacestrant: drug information. In: Lexicomp Online. Waltham, MA: UpToDate, Inc; 2023. https://online.lexi.com. Accessed June 15, 2023.
5. Rezlidhia (olutasidenib) product information. South San Francisco, CA: Rigel Pharmaceuticals, Inc; December 2022.
6. Pelcovits A, Niroula R. Acute myeloid leukemia: a review. R I Med J (2013). 2020;103(3):38-40.
7. Krazati (adagrasib) product information. San Diego, CA: Mirati Therapeutics, Inc; December 2022.
8. Alexander M, Kim SY, Cheng H. Update 2020: management of non-small cell lung cancer. Lung. 2020;198(6):897-907.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



