US Pharm. 2007;32(12)HS-3-HS-8.
Sickle cell disease (SCD) is an
all-encompassing term used to describe multiple hemoglobinopathy genotypes,
including sickle cell anemia (SCA), sickle-hemoglobin C disease, and
beta-thalassemia. The sickle cell trait is passed between generations in an
autosomal recessive process. Those individuals who are heterozygous for this
trait are considered carriers and normally will not show any clinical
manifestations of the disease, while those that are homozygous for the trait
have SCA.
Roughly 2 million Americans
are carriers of the sickle cell trait, while 72,000 suffer from sickle cell
anemia.1 Individuals of African, Mediterranean, Middle Eastern,
Indian, Caribbean, and South and Central American ancestry have the highest
rates for the sickle cell trait.2 In 1973, the average life
expectancy of a patient with sickle cell was 14 years. Secondary to advances
in ongoing disease management, therapy, and a comprehensive care models, the
average life span of a patient with sickle cell extended to 50 years in 2003.
3
Etiology
Sickle cell disease
was first described by a Chicago physician named James Herrick in 1910.
4,5 It was not until 1945 that the disease process was hypothesized as a
consequence of a hemoglobin abnormality.5 Normal hemoglobin (Hb-A)
is composed of two alpha and two beta globin chains.6 Sickle
hemoglobin (Hb-S) varies from Hb-A by a single amino acid mutation
substitution between valine and glutamic acid in the sixth position on the
beta globin chains.4
Pathophysiology
When deoxygenation
(decreased O2 tension) occurs, the hemoglobin molecule undergoes
rearrangement secondary to the amino acid substitution. The red blood cell
becomes elongated, rigid, and curved on the ends, resembling a sickle shape.
7 Abnormalities within the red blood cells are not limited to the
hemoglobin; intracellular potassium is rapidly decreased, and abnormalities
exist in cellular membrane phosphorylation as well as in the calcium pump.
4 These abnormalities can lead to dehydration within the cell and
increased membrane calcium levels, leaving the cell in an irreversible sickle
shape.8
Diagnosis
Although the
disease does not present clinically until four or five months after birth, the
Hb-S is present in the blood stream at birth. Umbilical cord blood samples can
undergo electrophoresis; for diagnosis of SCA the sample must be homozygous
for Hb-S. All babies from ethnic groups at high risk for SCA should be
screened for the disease at birth, because this has shown to decrease
mortality of very young children when the diagnosis is confirmed.4
Other tests can be performed for diagnosis; however, these do not
differentiate between the genotypes mentioned earlier.
Clinical Presentation
Infants have a
large percentage of fetal hemoglobin (Hb-F). Hb-F has a higher affinity for
oxygen compared to Hb-A and sickles less than Hb-S.8 With a low
propensity to sickle, increased levels of Hb-F have been shown to improve the
clinical course of the disease. As the infant's red blood cells begin to turn
over, Hb-S replace the Hb-F. The classic signs of SCA are red blood cell
destruction (hemolysis) and episodic vaso-occlusive crises. Hemolysis causes
the sickle red blood cell to have a decreased lifespan of four to 20 days,
while that of a normal red blood cell is 120 days.1 Sickle cells
can occlude the vasculature, ultimately leading to chronic organ damage (
TABLE 1).
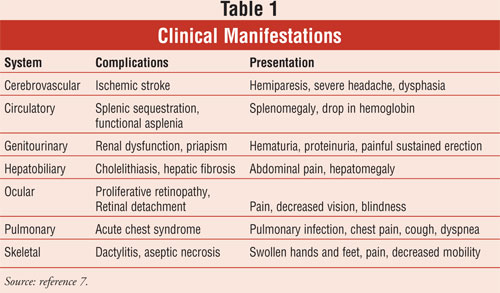
Clinical Manifestations
Vaso-occlusive
crises are complex in nature, resulting from interactions between endothelium,
plasma factors, leukocytes, and sickle cells.4 These complex
interactions result in tissue hypoxia, tissue death, and pain. Vaso-occlusive
episodes are the hallmark of SCA and occurrences range from daily to yearly.
The most common sites for vaso-occlusive crises are the skeletal, pulmonary,
and digestive systems, and a primary concern is resulting chronic organ
damage. By age 50 nearly half of patients with SCA are diagnosed, with at
least one chronic organ failure syndrome.9 Close monitoring and
follow-up of pulmonary, renal, and cardiovascular systems by a
multidisciplinary health care team are necessary to help slow the progression
of chronic organ damage. The following sections will discuss the common
clinical manifestations of SCA in the general population.
Cerebrovascular:
Stroke is one of the most devastating consequences of SCA, particularly in
childhood. By age 20, 11% of individuals with SCA will have suffered a stroke.
10 The Stroke Prevention Trial in Sickle Cell Anemia (STOP) was a
randomized study to determine the benefit of chronic red blood cell
transfusions in prevention of first stroke in high-risk children identified as
those with abnormal transcranial Doppler (TCD) velocities, the gold standard
for determining risk.10,11 First stroke was reduced by 92% in the
chronic transfusion group, and the study was terminated prematurely secondary
to the significant benefit. In response to this study, a clinical alert was
sent out recommending that all children with abnormal TCD velocities receive
chronic transfusion therapy for primary prophylaxis of stroke. It is now
recommended that children 2 years and older undergo annual TCD screening in
addition to close monitoring by health care providers for any changes noted in
neurocognitive function.3
Circulatory:
Tissue hypoxia in the spleen presents as abdominal pain and is so common that
by age 8 the spleen shrinks in size secondary to scarring (autosplenectomy).
4 The primary concern with this condition is the resulting immune
dysfunction and increased risk of infection, particularly from encapsulated
organisms, such as Streptococcus pneumoniae, Haemophilus influenza
.3 Decreased mortality in young children resulted from the use of
prophylactic penicillin to the age of 59 months (TABLE 2).3,4
Immunizations are also an important component to preventing infections. In
addition to the other required vaccines, children with SCA should also receive
the pneumococcal vaccine.6

Treatment
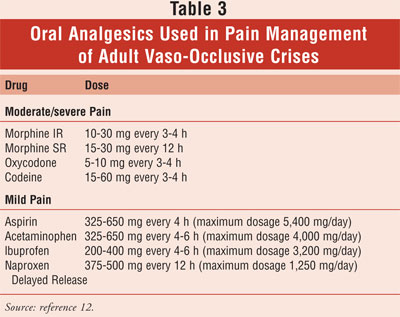
Pain
Management:Pain in patients with SCA has been difficult to manage
because there is no objective measurement parameter. Through the years,
management has improved with the advances of day centers for pain crises and a
better understanding of the disease process. Pain management is unique to each
patient and requires specific tailoring to previous successes and failures. A
list of commonly used oral analgesics is shown in TABLE 3. The biggest
barrier to properly controlling pain is the fear of addiction. Discussing the
difference between physical dependence, opioid tolerance, and addiction is
therefore paramount. Implementing a self-reporting pain log is one way to
assess a patient's pain and the efficacy of their current management.
Preventative Therapy
Transfusion
Therapy:Transfusion therapy is a mainstay in the treatment of patients
with SCA. Indications for transfusion therapy include acute chest syndrome,
heart failure, multiorgan failure syndrome, and stroke.3 Chronic
transfusion therapy has been studied in children for primary stroke
prophylaxis. The STOP II trial was implemented in the same study population to
determine the length of prophylaxis therapy. Unfortunately, the outcome of
this study showed that patients who had reached normal TCD velocities after
receiving chronic transfusion therapy that was halted after 30 months
eventually reverted back to high-risk velocities and risk for stroke.11
The appropriate length of chronic transfusion therapy is a controversial topic
and needs to be discussed on an individual basis between the patient and
health care provider.
Transfusions do not come
without risks, most commonly iron overload, infection, and alloimmunization.
Alloimmunization occurs when the recipient produces alloantibodies, making
future donor matching and transfusions more difficult.2 Therefore,
benefits of therapy must outweigh the risks. Prior to transfusing,
sickle-negative donor blood is phenotypically matched and leukocyte reduced
(to avoid febrile reactions).2 Two different types of transfusions
exist, simple and exchange. Simple transfusions deliver additional units of
blood without removal of sickle cells. This can increase blood viscosity and
is best reserved for when the hemoglobin level is less than 8 g/dL. Exchange
transfusions are more complicated and involve removing sickle cells and
replacing them with normal cells. This can be done manually or via automated
red cell exchange and is associated with decreased incidence of iron overload
and number of sickle cells.2,3 Exchange transfusions are warranted
if the hemoglobin level is high because it is not associated with an increase
in blood viscosity.
Iron overload (hemosiderosis) is the major
concern with chronic transfusion therapy and can lead to endocrine failure,
cirrhosis, and cardiomyopathy. Although serum ferritin levels are an accurate
source of iron stores, they are not accurate for assessing iron overload. Iron
overload is caused from an increased absorption of iron over an extended
period of time and leads to insoluble iron depositing in the visceral tissues.
Iron overload symptoms do not present until endocrinopathies and/or cardiac
failure exists.13 To properly assess iron overload, the iron dry
weight from liver biopsy should be used, with 7 mg/g dry weight as a
determinant for chelation therapy.2
Chelation therapy has been a leading cause
in decrease in quality of life for SCA patients. Until the recent approval of
an oral chelating agent, patients suffered from multiple injections, lost days
from work, pain, and sleep disturbances from administration pumps.13
Available chelating agents are summarized in TABLE 4. Monitoring of CBC
counts and renal function are recommended at baseline and throughout treatment
with deferasirox, whereas with deferoxamine therapy, auditory and
ophthalmologic exams should be monitored regularly.14

Hydroxyurea:
Hydroxyurea is a ribonuclease reductase inhibitor used in patients with SCA to
decrease vaso-occlusive episodes. Although its exact mechanism of action is
not known, it has shown to decrease the risk of sickling by increasing Hb-F
and red cell deformability, decreasing white blood cell counts, and altering
adhesive receptors on sickle reticulocytes and endothelium.1,5,15
The Multicenter Study of Hydroxyurea in Sickle Cell Anemia (MSH) demonstrated
that over 2.5 years, hydroxyurea reduced the morbidity of adult patients with
SCA by reducing the incidence of painful crises and ACS episodes by almost
half.15
Hydroxyurea is useful in its
ability to increase patients' quality of life and decrease morbidity. Many
factors need to be considered and evaluated prior to and during therapy (
TABLE 5). However, without the ability to retard the progression of
chronic organ damage, it is not the answer to sickle cell anemia.

Future Therapy Options
Butyrate is a
short-chain fatty acid that inhibits histone deactylase (HDAC) and increased
Hb-F production in animal models. Arginine butyrate, when administered to
patients (four days every four weeks), resulted in an induction of Hb-F in a
majority of patients. However, the large volume of drug needed and the need
for central catheters makes this drug difficult to administer. An oral dosage
form alternative is needed.5
Decitabine is an analog of
5-azacytidine (initial Hb-F inducer) that does not incorporate into RNA,
diminishing fears of carcinogenicity seen with 5-azacytidine. Small studies
have been performed with encouraging results. All patients who received
decitabine experienced Hb-F induction, regardless of previous hydroxyurea use.
Larger scale studies are needed to examine the full potential and safety of
this agent.5
Bone marrow transplantation is
the only curative therapy for SCA. It replaces the source of sickle cell
production and replaces it with bone marrow that produces normal cells. The
procedure is not without its risks; there is a 20% mortality associated with
the procedure. The inability to find a matched sibling donor has also limited
its use in treating SCA. Current research is evaluating the use of cord blood
transplantation as a means of increasing the amount of available stem cells
for transplantation.5
Sickle cell anemia is a
disease that currently is treated with supportive care, as no readily
available cure exists. The care of patients with SCA needs to be handled by a
multidisciplinary health care team, including pharmacists. Patients and their
caretakers have access to pharmacists on at least a monthly basis and often
need support in their challenging care plans. Pharmacists often serve as a
conduit for continuity of care between patients and their health care
providers. As drug experts, pharmacists can also lend their knowledge to the
complicated pain management and antibiotic regimens.
References
1. Dauphin-McKenzie N, Gilles JM,
Jacques E, et al. Sickle cell anemia in the female patient. Obstet Gynecol
Survey. 2006;61:343-352.
2. Management and
therapy of sickle cell disease. NIH Publication No. 02-2117 Revised June 2002
(Fourth Edition). National Heart, Lung,
and Blood Institute. Available at:
www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf. Accessed October 1,
2007.
3. Claster S, Vichinsky
EP. Managing sickle cell disease. BMJ. 2003;327:1151-1155.
4. Ranney HM, Sharma V.
Disorders of hemoglobin structure. In: Beutler E, Lichtman MH, Coller BS, et
al, eds. Williams Hematology. New York, NY; McGraw-Hill; 2001:345-354.
5. Frenette PS, Atweh
GF. Sickle cell disease: old discoveries, new concepts, and future promise.
J Clin Invest. 2007;117:850-858.
6. Wilson RE,
Krishnamurti L, Kamat D. Management of sickle cell disease in primary care.
Clin Pediatr. 2003;42:753-761.
7. Baldy CM. Red blood
cell disorders. In: Price SA, Wilson LM, eds. Pathophysiology: Clinical
Concept of Disease Processes. 5th ed. St. Louis, MO: Mosby-Year Book,
Inc.; 1997:197-206.
8. Curry CE Jr, Beasley ED. Sickle
cell anemia. In: Dipiro JT, Talbert RL, Yee GC, et al., eds.
Pharmacotherapy: A Pathophysiologic Approach.4th ed. Stamford, CT:
Appleton & Lange; 1999:1573-1583.
9. Powars DR, Chan LS,
Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study
of 1056 patients. Medicine. 2005;84:
363-376.
10. Wang WC. The
pathophysiology, prevention, and treatment of stroke in sickle cell disease.
Curr Opin Hematol. 2007:191-197.
11. Lee MT, Piomelli S,
Granger S, et al. Stroke prevention trial in sickle cell anemia (STOP):
extended follow-up and final results. Blood. 2006;108:847-852.
12. Bauman TJ. Pain
management. In: Dipiro JT, Talbert RL, Yee GC, et al., eds.
Pharmacotherapy: A Pathophysiologic Approach. 4th ed. Stamford, CT:
Appleton & Lange; 1999:1020-1023.
13. Abetz L, Baladi JF,
Jones P, et al. The impact of iron overload and its treatment on quality of
life: results from a literature review. Health Qual Life Outcomes.
2006;4:73.
14. Stumpf JL.
Deferasirox. Am J Health-Syst Pharm. 2007;64:606-616.
15. Steinberg MH,
Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in
adult sickle cell anemia. JAMA. 2003;289:
1645-1651.
To comment on this article, contact
editor@uspharmacist.com.



