US Pharm. 2018;43(3):HS-10-HS-15.
ABSTRACT: Pulmonary hypertension (PH) is a hemodynamic and pathophysiologic condition that presents with abnormal elevation in pulmonary circulation pressure. Pulmonary arterial hypertension (PAH) is a progressive disease that affects the small pulmonary arteries and is characterized by vasoconstriction, medial hypertrophy, cell proliferation and fibrosis, complex lesions (plexiform lesions), and thrombosis in situ. Right-heart catheterization should always be performed to confirm the diagnosis of PAH and chronic thromboembolic PH. Currently there are FDA-approved treatment options for patients with PAH, including ambrisentan, bosentan, macitentan, epoprostenol, iloprost, treprostinil, selexipag, sildenafil, and tadalafil.
Pulmonary hypertension (PH) is a hemodynamic condition causing unstable cardiovascular and pulmonary indices that presents with abnormal elevation in pulmonary circulation pressure.1-3 Normal mean pulmonary arterial pressure (mPAP) is between 14 +/- 3 mmHg and 20 mmHg.1,3 PH is defined as an increase in mPAP greater than 25 mmHg at rest as assessed by right-heart catheterization (RHC).1-3 The clinical significance of an mPAP between 21 and 24 mmHg is unclear; however, patients presenting with an mPAP in this range should be monitored closely as they may be at risk for developing pulmonary arterial hypertension (PAH).
The clinical classification of PH categorizes PH into five groups (Table 1) with different pathological, pathophysiological, prognostic, and therapeutic characteristics.1,3 PAH is currently classified as Group 1.1,3 PAH, the focus of this article, involves a subset of PH patients characterized by the presence of precapillary PH.1,3
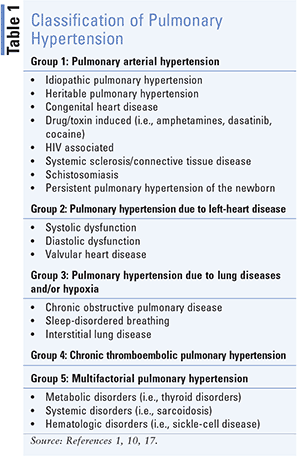
PAH may occur in isolation or along with other clinical conditions (Table 1).1-3 PAH is classified as idiopathic (IPAH), heritable (HPAH), or drug- and toxin-induced, and is associated with connective tissue disease, HIV infection, portal hypertension, congenital heart disease, or schistosomiasis.1,3 In pediatrics, persistent PH of the newborn is also a part of Group 1 PH. There are several registries that describe the epidemiology of PAH.1 The estimates of incidence and prevalence for PAH were 2.4 cases per million adults and 15 cases per million adults, respectively.1,3 The estimate for prevalence of idiopathic PAH is 5.9 cases per million adults.1
PATHOPHYSIOLOGY
PAH is a progressive disease that affects the small pulmonary arteries (<50 microns) and is characterized by vasoconstriction, medial hypertrophy, cell proliferation and fibrosis, complex lesions (plexiform lesions), and thrombosis in situ.4 Pulmonary arterial changes occur in all three layers of the pulmonary artery: the tunica adventitia, tunica media, and tunica intima. The intima comprises endothelial cells that line the lumen. The media consists of smooth muscle cells, and the adventitia consists of collagen fibers and fibroblasts. The internal elastic lamina separates the intima from the media, and the external elastic lamina separates the media from the adventitia. Some key pathobiologic changes in PAH include vasoconstriction, arterial remodeling/inflammation, plexiform lesion, and thrombotic lesion.
Vasoconstriction results in the narrowing of the lumen and tightly folded internal elastic lamina with endothelial cells pinched between the folds. Arterial remodeling and inflammation causes thickening of the adventitia and media with neointima formation due to smooth cell and fibroblast proliferation and migration, and lymphoid neogenesis. Plexiform lesions are glomeruloid-like lesions resulting from aberrant angiogenesis; thrombotic lesions are recanalized thrombus in the lumen.3 Prolonged pulmonary vasoconstriction leads to endothelial dysfunction, which is characterized by increased levels of vasoconstrictors (endothelin) and reduced production of vasodilators (nitric oxide and prostacyclin).3 These mechanisms have led to the development of pharmacologic agents that counteract factors involved in the pathogenesis of PAH. IPAH and HPAH are rare and have been associated with predisposing genetic factors such as mutations in the bone morphogenetic protein receptor type II that is involved in the control of vascular cell proliferation.
Diagnosis/Clinical Manifestations
PH is a multifaceted group of disorders resulting from various pathophysiologic and hemodynamic mechanisms with nonspecific symptoms, usually due to other underlying comorbid conditions.5 This causes the diagnosis to be particularly challenging, necessitating a stepwise approach. History and physical examination are usually evaluated first. A history of angina, dyspnea on exertion, exercise intolerance, fatigue, and syncope are among some of the signs that, although not exclusively related, can help support differential diagnosis. Upon physical examination, abnormal pulse oximetry, tricuspid regurgitation, lower-extremity edema, and signs of right-heart failure can be a few of the signs suggestive of PH.5 Initially, a noninvasive test such as an echocardiograph can be performed in patients presenting with any of these signs. Echocardiography or cardiac echo is simply a sonogram of the heart that provides information on abnormalities.5-7 This should always be performed when PH is suspected. An estimated systolic pulmonary arterial pressure of 35 to 40 mmHg is suggestive of PH and may prompt further testing.5 Among the five PH groups, all can be defined as an increase in mPAP ≥25 mmHg at rest upon RHC based on the 2015 European Society of Cardiology/European Respiratory Society joint task force guidelines.1
RHC is an invasive test that commonly can be completed in about an hour in an outpatient setting. A thin catheter is inserted into a vein of the groin or neck, advancing into the heart. This catheter takes pressure measurements in the various heart chambers and pulmonary arteries and measures oxygen in the blood and cardiac output. The RHC should always be performed to confirm the diagnosis of PAH and chronic thromboembolic pulmonary hypertension (CTEPH), and when considering treatment of PH.18,9 The various PH groups may differ diagnostically based on RHC measurements. PAH is diagnosed based on an RHC mPAP >25, pulmonary artery wedge pressure ≤15 mmHg, and peripheral vascular resistance >3 Wood units, without other causes of PH.1
Vasoreactivity testing is only used for identifying patients who qualify for high-dose calcium channel blocker (CCB) therapy. Patients with IPAH, HPAH, or drug-induced PH are suitable for this test. Inhaled nitric oxide, IV epoprostenol, IV adenosine, or inhaled iloprost may be employed, with inhaled nitric oxide the preferred standard of care. A positive response is defined as reduction of mPAP ≥10mmHg to reach an absolute value of mPAP ≤40 mmHg with an increased or unchanged cardiac output.1,5 After appropriate diagnosis and evaluation, treatment can be considered.1
MANAGEMENT
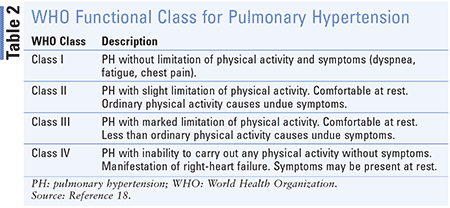
Treatment options for PH depend on underlying etiology, WHO functional class, and/or severity of symptoms. (Tables 2-4). The goals of treatment are to achieve low-risk status, which is associated with good exercise capacity, improved quality of life, good right-ventricular function, and low mortality risk.1 Current therapies work by causing and/or targeting pulmonary artery vasodilation through various mechanisms. Other PH treatments with vasodilator therapy are not recommended and/or may cause harm or worsening of disease.5 None of the targeted therapies have been shown to slow pathological progression.10
In addition to targeted therapy for vasodilation, supportive therapy is necessary. While not the focus of this article, the pharmacist should not forget that appropriate management of complications such as arrhythmias, hemoptysis, and mechanical complications may be needed.1 It is important to regularly assess PAH patients. This assessment should include checking for evidence of clinical worsening, progression of PAH, stability of right-ventricular function, and the patient’s current status associated with prognosis. Therefore, checking hemodynamic status and complications of disease is recommended. These factors can help drive modifications of pharmacotherapy and other interventions.
SUPPORTIVE THERAPY
Oral anticoagulation, diuretics, oxygen, digoxin, and management of anemia may be necessary in supportive management of patients with PAH. Venous thromboembolism in certain forms of PH is a possibility because of abnormalities in coagulation and fibrinolytic pathways along with right-heart failure and immobility.1 This makes anticoagulation recommended for IPAH, HPAH, CTEPH, and PAH. Diuretic therapy may be considered in patients with symptoms of decompensated right-heart failure resulting in fluid retention, ascites, peripheral edema, and other related symptoms.1 Oxygen has been shown to reduce peripheral vascular resistance, but long-term therapy has not been shown to be beneficial. Oxygen may be recommended in patients with chronic obstructive pulmonary disease, low arterial blood-oxygen pressure, evidence of symptomatic benefit, and correctable desaturation on exercise, especially in patients with Group 3 chronic lung disease. Digoxin may improve cardiac output in IPAH and slow ventricular rate in patients with arterial tachyarrhythmias. Evidence of efficacy for chronic use in PH is lacking.1 Iron status should be monitored in patients with PH because iron deficiency may reduce exercise capacity and increase mortality. Since iron deficiency is common in this population, iron supplementation can be recommended once evaluation is completed. Theoretically, IV iron replacement is usually preferred over oral therapy because oral therapy is associated with impaired absorption in these patients.1
TARGETED/SPECIFIC DRUG THERAPY
Calcium Channel Blockers
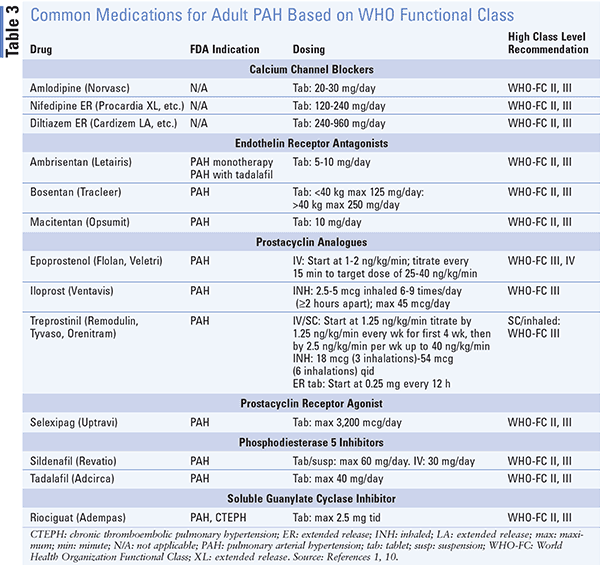
CCBs inhibit arterial calcium channels.10 A trial of CCBs is only recommended for patients with a positive response to vasoreactivity testing on RHC.1 The most commonly used CCBs are nifedipine, amlodipine, and diltiazem. Determining the baseline heart rate is necessary when choosing an appropriate CCB. If a patient has bradycardia, nifedipine or amlodipine should be used. If a patient has tachycardia, diltiazem is chosen. High dosages are usually needed to treat this population. IPAH patients receiving CCB therapy should be reassessed after 3 to 4 months of therapy. It is important to note that CCB therapy can lose effectiveness over time.1,10 Upon evidence that effectiveness is waning, alternative therapies may be recommended.
Endothelin Receptor Antagonists
Endothelin is a peptide or compound that constricts blood vessels and raises blood pressure. There are three identified forms of endothelin, which vary in the regions of expression and binding of receptors. Endothelin-1 is most implicated in PAH pathogenesis. Endothelin-1 exerts effects through vasoconstriction by binding to two distinct receptors, type A and B, in the pulmonary vascular smooth muscle. Endothelin type A (ETA) is located on vascular smooth muscle cells causing vasoconstriction, cell proliferation, and proinflammatory effects. Endothelin type B (ETB) has two subtypes; ETB1 is located on endothelial cells and causes nitrous oxide–mediated vasodilation, whereas ETB2 causes vasoconstriction on vascular smooth muscle.1,11
Endothelin receptor antagonists (ERAs) are a class of drugs that block the ETA and ETB endothelin receptors; their selectivity varies.11 These medications also block vascular hypertrophy, fibrosis, and inflammation, all possible mechanisms in the PAH disease process.4,10
There are currently three available FDA-approved ERAs—ambrisentan, bosentan, and macitentan. Ambrisentan preferentially binds to ETA.10 Bosentan and macitentan bind to ETA and ETB. All ERAs improve exercise capacity and delay clinical worsening. Macitentan has been shown to reduce hospitalizations, and ambrisentan in combination with tadalafil has been shown to reduce risk of clinical progression of disease along with reducing hospitalizations.10 This class of drugs is teratogenic, limited to restricted distribution through Risk Evaluation and Mitigation Strategies programs, and only available through specialty pharmacies. These drugs may also cause peripheral edema and/or pulmonary edema. Bosentan, the original drug in this class, interacts with oral contraceptives, statins, ketoconazole, amiodarone, and glyburide, among others. Ambrisentan is the only drug that does not cause major CYP450 interactions. These drugs are not recommended in patients with moderate-to-severe liver disease; therefore, liver function tests should be monitored at baseline and as clinically indicated, depending on the drug. It is recommend that hemoglobin be measured and monitored at baseline, monthly, and periodically as clinically indicated, with more frequent monitoring based on the drug.10
Prostacyclin Analogues/Prostacyclin-Receptor Agonists
Prostacyclin induces potent vasodilation of all vascular beds. It is also found in endothelial cells. Prostacyclins also act as potent platelet-aggregation inhibitors and may have cytoprotective and antiproliferative effects. Prostacyclin analogues/prostacyclin-receptor agonists are beneficial for PAH patients because they address the disrupted prostacyclin pathways found in some pulmonary arteries.1,10,12,13 There are currently three FDA-approved prostacyclin analogues: epoprostenol, iloprost, and treprostinil. Epoprostenol and treprostinil improve exercise capacity, while iloprost improves symptoms and exercise tolerance, and stabilizes clinical disease progression. Selexipag is the only FDA-approved prostacyclin-receptor agonist. Selexipag can delay clinical progression and reduce the risk of hospitalization. The prostacyclin analogues may cause bleeding due to platelet aggregation inhibition. There have been reports of hyperthyroidism with epoprostenol and selixipag.
All prostacyclin-related drugs are only available through specialty pharmacies.10 Epoprostenol is given as a continuous infusion and requires central access for long-term use. Iloprost is available as inhalation therapy only. Treprostinil is available in three different formulations: for injection (SC and IV); inhalation; and as orally administered extended-release tablets. All formulations are available under various brand names. Selixipag is only available as a tablet and has strong CYP interactions. Most of these prostacyclin-related medications commonly cause headache, nausea, flushing, and jaw pain.10 These drugs are titrated upward to achieve appropriate clinical response.
Phosphodiesterase (PDE)-5 Inhibitors
This class of drugs inhibits the PDE-5–enhancing effects of nitric oxide-activated increases in cyclic guanosine monophosphate, causing vasodilation.10,14 These drugs were approved for erectile dysfunction but have become popular in the treatment of PAH. Although most of these medications have been studied for this population, the only FDA-approved PDE-5 inhibitors for PAH are sildenafil and tadalafil, available under different brand names than their erectile-dysfunction counterparts.10 These drugs should be avoided with nitrates and strong CYP 3A4 inhibitors. Grapefruit may increase concentrations of these drugs. Both medications are available in oral tablet and/or solution form.10
Soluble Guanylate Cyclase Inhibitor
Riociguat is the only FDA-approved soluble guanylate cyclase inhibitor that stimulates the receptor for nitrous oxide through cyclic guanosine monophosphate production.10,15,16 This drug improves exercise capacity, improves WHO functional class, and delays clinical worsening. It is contraindicated with concomitant use of nitrates or PDE-5 inhibitors. Appropriate discontinuation of PDE-5 inhibitors before stopping riociguat is necessary to avoid complications from their interaction. At least 24 to 48 hours are needed for transitioning between these therapies, depending on which drugs are being used. Common side effects are bleeding and hypotension. Dose reduction is needed when used with strong CYP and P-glycoprotein inhibitors.10 One caveat is that smokers may requires higher dosages when using riociguat; if the patient stops smoking, the drug doses may need to be lowered.1,10 Riociguat is only available as a tablet, and like the ERAs, this drug is teratogenic and only available through specialty pharmacies.10 Studies have shown favorable benefit-versus-risk in CTEPH patients.16 Riociguat is also the only drug approved by the FDA for both PAH and CTEPH.10,16
THERAPY SELECTION
Depending on the severity of the disease, two or more drugs from different classes are usually needed to treat PAH.1,10 Oral drugs, used as monotherapy or in combination, are recommended for less severe disease; parenteral prostacyclins are usually used as combination therapy for more severe disease. Owing to the more stable effects of parenteral prostacyclins, these are preferred over inhaled versions.10 When considering combination therapy, options include either sequential or initial therapy. As the most commonly used combination therapy, sequential therapy is started with monotherapy and a second or third drug is added based on inadequate response or clinical deterioration. Initial or upfront combination therapy—that is, beginning with more than one drug—is used when there are increased mortality risks or concurrent severe medical illnesses based on WHO functional class.1 Surgical procedures, such as balloon atrial septostomy, demonstrate improved left-ventricular preload, cardiac output, and oxygenation. These procedures may be considered and provide benefit for patients who are WHO functional class IV with right-heart failure refractory to standard medical therapy or with severe syncopal symptoms. Lung and heart-lung transplant, pending organ availability, remain viable options for consideration after patients have demonstrated an inadequate response to maximal combination therapy.1
SPECIAL POPULATIONS
Women’s Health
There are many factors to consider regarding the health of women with PAH, such as pregnancy status and hormonal aspects. Unfortunately, pregnancy is associated with increased mortality in PAH; however, studies have shown that when PAH is well controlled, the outcomes are more favorable.1 Effective contraception is recommended, especially when teratogenic agents are being used. Barrier contraception, although safe for a PAH patient’s health, is unpredictable at preventing pregnancy. Progesterone-only contraception is recommended, such as the traditional mini-pill or contraceptive implants. If unintended pregnancy does occur, the patient should be informed of the high risk and termination should be discussed. If the pregnancy is continued, targeted therapy, planned elective delivery, and appropriate consultation with the necessary specialists should occur. Currently it is not known whether hormonal therapy for postmenopausal women with PAH is recommended. If needed, appropriate anticoagulation should be used.1
Pediatrics
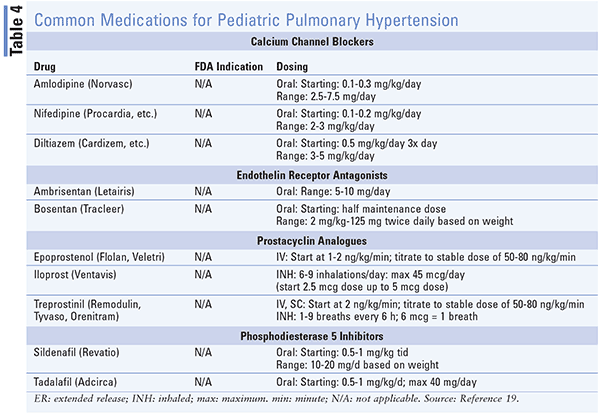
PH can occur at any age from birth throughout childhood. There are unique features of the condition in this population that are beyond the scope of this article. IPAH, HPAH, and congenital heart disease PAH are the most common types of PH in pediatrics.1 Evaluation and diagnosis remains the same as in adults, with vasoreactivity testing on RHC being confirmatory. Importantly, there is evidence that heart catheterization carries greater risk in children than in adults, especially in children aged less than 1 year. The same treatment modalities (Table 4) and algorithms used in adults may be used with children, in particular PDE-5 inhibitors and ERAs.1
ROLE OF THE PHARMACIST
The complexities of PAH and the many drug therapies available for treatment calls for the involvement of the clinical pharmacist, directly or indirectly, in the care of these patients. Pharmacists can provide their expertise for this population in ambulatory, hospital, specialty pharmacy, and managed-care settings. Because most of the drugs for PAH are classified as specialty medications, owing to their great expense, sensitive administration, and intensive clinical monitoring, pharmacists are in a perfect position to ensure that the most cost-effective and appropriate therapies are being used.
In addition, the multitude of drug interactions and side effects that may occur with many of these agents can be identified and addressed. Helping to choose and recommend appropriate supportive and target-specific therapy for individuals and large populations being treated based on current clinical guidelines and other evidence-based protocols is another role for the pharmacist.
When considering dose titrations, these drugs must be handled carefully to avoid unwanted side effects. Clinical pharmacists can ensure that needed drugs are titrated appropriately based on response and adverse effects. Educating patients and prescribers on appropriate treatments, expectations, and the importance of adherence is essential. The assessment of functional severity shown in Table 2 can be used by pharmacists in assisting prescribers in identifying patients’ class severity. Clinical pharmacists should always consider all sources of information in order to understand all of the nuances of these complex regimens, owing to clinical variations among these drugs and drug classes that are beyond the scope of this review.
CONCLUSION
PAH is a progressive disease that affects the small pulmonary arteries; diagnosis is challenging. Current medication therapies work by causing and/or targeting pulmonary-artery vasodilation; supportive therapy is also necessary. PAH treatment for women and children requires special consideration. The complexities of PAH and the many drug therapies available for treatment call for the involvement of the clinical pharmacist in the care of these patients.
REFERENCES
1. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2016;37:67-119.
2. Pesto S, Begic Z, Prevljak S, et al. Pulmonary hypertension—new trends of diagnosis and therapy. Med Arch. 2016;70(4):303-307.
3. McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardio. 2015;65:1976-1997.
4. Rubin LJ, Hopkins W. UpToDate. Updated November 17, 2017. www.uptodate.com/contents/the-epidemiology-and-pathogenesis-of-pulmonary-arterial-hypertension-group-1. Accessed December 1, 2017.
5. Dunlap B, Weyer G. Pulmonary hypertension: diagnosis and treatment. Am Fam Physician. 2016 ;94(6):463-469.
6. Nihoyannopoulos P. Why is a new journal dedicated to echocardiography required? Echo Res Pract. 2014;1(1):E1–E3.
7. American Heart Association. What is echocardiography? www.heart.org/idc/groups/heart-public/@wcm/@hcm/documents/downloadable/ucm_300438.pdf. Accessed December 1, 2017.
8. Preston I, Rosenkranz S. Right heart catherization: best practice and pitfalls in pulmonary hypertension. Eur Respir Rev. 2015;24:642-652.
9. Right heart catheterization. Pulmonary Hypertension R.N. http://pulmonaryhypertensionrn.com/right-heart-catheterization-rhc/. Accessed December 1, 2017.
10. Meds for pulmonary arterial hypertension. Pharmacist’s Letter/Prescriber’s Letter. March 2017.
11. Kowalczyk A, Kleniewska P, Kolodziejczyk M, et. al. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arc. Immunol. Ther. Exp. 2015;63:41-52.
12. Mitchell J, Ahmetaj-Shala B, Kirkby N, et. al. Role of prostacyclin in pulmonary hypertension. Glob Cardiol Sci Pract. 2014; 2014(4):382-393.
13. Zhang H, Li X, Huang H, et al. Comparative efficacy and safety of prostacyclin analogs for pulmonary arterial hypertension. Medicine (Baltimore). 2016;95(4):e2575.
14. Unegbu C, Noje C, Coulson J, et. al. Pulmonary hypertension therapy and a systematic review of efficacy and safety of PDE-5 inhibitors. Pediatrics. 2017;139(3):e20161450.
15. Ghofrani HA, Galie N, Grimminger F, et. al. Riociguat for pulmonary arterial hypertension. N Engl J Med. 2013;369:330-340.
16. Simonneau G, D’Armini A, Ghofrani HA, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2). Eur Respir J. 2015;45:1293-1302.
17. Ryan J, Thenappan T, Luo N. The WHO classification of pulmonary hypertension: a case-based imaging compendium. Pulm Circ. 2012;2(1):107-112.
18. Popa A. Bosentan (Tracleer), a new agent for the treatment of pulmonary arterial hypertension. Cleveland Clinic Center for Continuing Education. Pharmacotherapy update. Volume V, Number 2. March/April 2002. www.clevelandclinicmeded.com/medicalpubs/pharmacy/marapr2002/bosentan.htm. Accessed December 1, 2017.
19. Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation. Epub November 3, 2015. http://circ.ahajournals.org/content/132/21/2037.long. Accessed December 1, 2017.
To comment on this article, contact rdavidson@uspharmacist.com.


