US Pharm. 2018;43(3)12-18.
ABSTRACT: Sickle cell disease is the most common inherited blood disorder, affecting millions of people worldwide. Although sickle cell disease can be found in various areas of the world, it predominantly affects individuals of African or Hispanic heritage. The hallmark clinical symptoms are due to vaso-occlusion, which occurs when the blood vessels are obstructed by sickled red cells. The most common symptom of vaso-occlusion is moderate-to-severe pain. Severe complications include acute chest syndrome, stroke, and pneumonia. Clinicians and patients with sickle cell disease may have difficulty treating the painful episodes owing to the subjective nature of pain and lack of knowledge of the disease.
Sickle cell disease is a blood disorder caused by inheriting two abnormal hemoglobin genes. Hemoglobin S is a mutated version of the normal hemoglobin gene A expressed in children and adults.1,2 The abnormal hemoglobin exhibits solubility and structural differences that lead to irregularly shaped red blood cells, typically in a sickle shape.1,2 These sickled blood cells interrupt the normal flow of blood, causing vaso-occlusion and decreased oxygen delivery to tissues and leading to pain, tissue damage, and tissue death (Figure 1).1,2 Complications from vaso-occlusion constitute the most common cause of death in patients with sickle cell disease.1,2
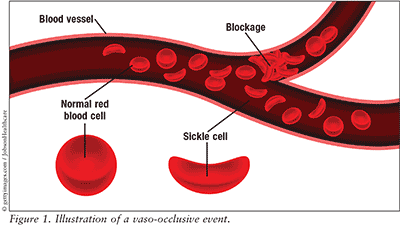
In patients with sickle cell disease, vaso-occlusive crisis is the leading cause of emergency department visits and hospitalization. Severe pain can be experienced as early age 6 months and throughout a patient’s life.1 Patients have a normal, healthy appearance with no external distinguishing clinical features. The pain most commonly occurs in the chest, back, and extremities and may last for multiple days or weeks.2 Knowing how to manage pain during a vaso-occlusive crisis is essential for patients with sickle cell disease to maintain a healthy lifestyle. Arguably, the most difficult aspect of treating sickle cell crises is assessing the patient’s pain level. Because pain is subjective and clinicians cannot precisely ascertain the severity of the patient’s pain, effective treatment often entails aggressive fluid hydration and IV opioid therapy.1
Epidemiology
In the United States, approximately 100,000 Americans have sickle cell disease. The disease is an autosomal recessive disorder that is genetically acquired from two abnormal alleles.1,2 People with sickle cell trait acquire one abnormal heterozygous allele, in contrast to sickle cell disease patients, who have two homozygous alleles. Among African Americans, 1 out of every 13 infants possesses the sickle cell trait and sickle cell disease occurs in 1 out of every 365 births.3 Every year, 1,200 to 1,400 infants in the U.S. are born with sickle cell disease. During the 1970s, the average life expectancy of a patient with sickle cell disease in America was less than age 20 years; however, due to innovative research and new medical treatments, the life expectancy has nearly tripled; the median life expectancy in sickle cell disease patients in 2014 was age 58 years.4
Pathophysiology
The clinical manifestation of sickle cell disease occurs due to a single base substitution found in the beta-globin gene located on chromosome 11; the mutation causes a substitution of glutamic acid with valine, leading to downstream production of the altered hemoglobin S.1,2 Deoxygenation of sickle hemoglobin causes a hydrophobic interaction with other hemoglobin molecules, producing aggregation of the hemoglobin. The polymerization of the sickle hemoglobin results in the distortion of the red cell's shape, which ultimately triggers the vaso-occlusive events. The rate and extent of polymerization formation depends on three factors: the cell’s degree of deoxygenation, the intracellular concentration of hemoglobin S, and the presence or absence of hemoglobin F. Cell dehydration and increased amounts of hemoglobin S enhance polymerization, while hemoglobin F inhibits polymerization by binding to glutamine.5 The sickled red blood cell’s life span decreases from the normal survival span of 120 days to 15 days.6
The molecular interaction of sickled red cells and the vascular endothelium also plays a critical role in the blood disorder’s pathogenesis. The adherence of red cells to the endothelium causes vascular injury, triggering inflammatory and coagulation cascades.1,2 Adhesion of the sickled cells to the endothelial layer is facilitated by the interaction of the vascular cell adhesion molecule stimulated by tumor necrosis factor-alpha. Additionally, the number of immature red blood cells or reticulocytes is increased in patients with sickle cell disease due to increased hematopoiesis; these cells have an integrin complex that binds to various molecules, such as fibronectin, endothelial cells, thrombospondin, and vascular-cell adhesion molecule, to increase occlusion of the vessels.4
Risk Factors and Patient Barriers
Due to the genetic nature of the disease, the blood disorder has no notable risk factors other than inheritance. The frequency of pain crises correlates positively with various factors. For instance, most pain episodes are reported to occur in October, November, and January due to cold temperatures.7 Cold weather may cause a crisis by decreasing the flow of blood and forming a blockade of sickled red cells in the arteries.1-3
Pregnant patients with sickle cell disease are also more likely to have a vaso-occlusive event. In addition to elevated crisis susceptibility, women with sickle cell disease are at increased risk for pregnancy complications such as low birth weight and preterm labor.8
The most important hematologic factors include hematocrit level and fetal hemoglobin level. At birth, the newborn infant’s red blood cells contain mostly fetal hemoglobin—hemoglobin F—which binds to oxygen with greater affinity than adult hemoglobin does. After birth, the gene expression changes to promote the production of hemoglobin A, adult hemoglobin. In children with sickle cell anemia, production of the defective hemoglobin S begins to occur and continues into adulthood.9 Consequently, by age 3 to 6 months, the infant with sickle cell disease will have red blood cells containing hemoglobin S and be at risk for vaso-occlusion.
Studies have shown that young adult patients between the ages of 20 and 29 years, especially males, have the highest rate of pain episodes.10 Patients in their 20s seem to have a relatively low fetal hemoglobin level, as well as a low hematocrit concentration. The combination of the two undesirable blood features produces low blood oxygenation, which may trigger a pain crisis.1-3
In addition to the risk factors mentioned above, patient and medical barriers can influence a vaso-occlusive event by preventing patients from receiving adequate treatment. Arguably, the most concerning barrier is the subjective nature of pain. The age and ethnic background of patients with sickle cell disease may contribute to biases in pain evaluation.11 Because pain cannot be directly assessed objectively, some clinicians may not fully appreciate patients’ reports of pain intensity and the amount of medication required. Sickle cell disease patients often know what works for them; therefore, a request for a specific drug or dose ought not to be interpreted as drug-seeking behavior.1 Among African Americans and other racial minorities, social stigma also plays a role in the treatment of sickle cell disease.11 According to Jenerette and Brewer, stigmatization may hinder patients with sickle cell disease seeking care for acute pain exacerbations, especially during a young adult’s transition from pediatric care to adult care.11
Clinical Features and Symptoms
Due to an elevated fetal hemoglobin level at birth, patients typically do not exhibit signs of sickle cell disease until 6 months after birth. Other than the manifestation of vaso-occlusion, common symptoms of sickle cell disease encompass such characteristics of anemia as fatigue, exercise intolerance, and shortness of breath. Pediatric patients may experience delayed growth and fever, while adults may present with ulcers or bone injury. Other symptoms include jaundice, frequent urination, and muscle weakness.1-3
Key clinical features of sickle cell disease involve microvascular disruption of organs. A vaso-occlusive event can cause various complications, potentially resulting in permanent damage of vital organs such as the lungs, heart, kidneys, spleen, and brain. Common complications and leading causes of death are acute chest syndrome and stroke.12 Due to splenic dysfunction, sickle cell disease patients have a weakened immune system; they are more susceptible to infections and may need antibiotic treatment for prophylaxis. Other complications that may arise include priapism in men, pneumonia, and retinopathy.1-3
Pain Assessment
Effective pain management requires frequent reassessments of pain to achieve pain relief. Adjustments to a pain regimen may be needed to manage opioid tolerance, opioid side effects, and, primarily, to achieve pain control. The subjective nature of pain may make an assessment of the severity of a patient’s vaso-occlusive crisis difficult, but published guidelines for health professionals have provided recommendations on evaluating a sickle cell patient’s health status to optimize treatment regimens.1,2 With the exception of infants, clinicians should look to patients’ self-reports as the primary source of assessment.13
Self-reports may be supplemented with physical findings, laboratory data, and diagnostic information. Frequency of pain episodes, age, chronic comorbidities, functional status, emotional health, and cognitive ability should be considered in patient assessment.1 Laboratory values such as oxygen saturation, blood pressure, and temperature should be noted as well in deducing the severity of the clinical situation.13 After the clinician selects a method of measurement for pain intensity, the measurement should be recorded and the patients should be reassessed frequently for appropriate treatment. Pain relief should be assessed after each treatment regimen and when sufficient time has elapsed for the drug to reach optimal efficacy.
The instruments most commonly used to assess pain are numeric scales, in which clinicians ask patients to report their pain level as a number from zero to 10. A score of zero indicates no pain, whereas a score of 10 signifies the worst possible pain. For children who are unable to understand numbers or for non–English speaking patients, physicians may use picture pain scales such as the Oucher scale or Wong-Baker scale to assess pain intensity.1 The clinician should explain that each picture of a face depicts a person who is happy because he or she has no pain or sad because he or she has a lot of pain.1
Nonpharmacologic Management
Regardless of the individual’s state of hydration, fluids are routinely administered to patients experiencing acute pain episodes. The goal of hydration is to slow or stop the sickling process by increasing the blood plasma volume, which indirectly reduces red-cell dehydration and the intracellular concentration of deoxygenated hemoglobin S. In children and adolescents, between one and one-and- a-half times the daily estimated fluid maintenance, or 3 to 4 L, of fluids is recommended.13,14 In adults, studies recommend that IV fluid replacement be administered at a rate of 250 mL/hour and then reduced to 125 mL/hour if there are no signs of congestive heart failure or renal failure.14 Oral hydration should be supplemented with IV fluids to further improve blood flow. Even for patients with sickle cell disease with no recent acute episodes, fluid hydration is essential for maintaining a healthy lifestyle.
Blood transfusions deliver donor erythrocytes that contain red blood cells with normal hemoglobin A, reducing the percentage of circulating red blood cells in hemoglobin S. Two methods of infusion are simple transfusion and exchange transfusion. A simple transfusion infuses red blood cells into the recipient without removing of the patient’s blood, whereas an exchange transfusion replaces the patient’s blood before or during the infusion. In addition to increasing hemoglobin A levels, exchange transfusions increase the recipient’s overall blood volume without increasing the viscosity. Prominent indications for blood transfusions include protection of organ function from sickled erythrocytes and prevention of strokes. Although blood transfusions may be beneficial in preventing and treating vaso-occlusive crises, the therapy has some risks that are amplified by sickle cell disease.1,2,13 These risks include alloimmunization (antibody development that can lead to increased risk of transfusion reactions and red blood cell lysis), iron overload, and the frequent need for venous access.2 Because of the risks, blood transfusion is usually used in patients with severe complications or severe anemia (as indicated by severely low hemoglobin); when this method is employed, posttransfusion hematocrit should be less than 36%.1
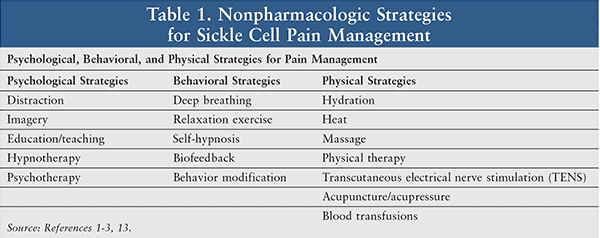
Other nonpharmacologic methods of pain management, including psychological and behavioral strategies, are listed in Table 1.
Pharmacologic Management
Analgesics are the foundation of the management of sickle cell pain (Tables 2, 3). During a vaso-occlusive episode, aggressive IV opioid therapy should be initiated.1-3,13 Opioids should be selected based on the intensity and duration of the pain. Opioids such as hydromorphone and morphine are usually selected because of their quick onset of action and the multiple dosage forms available. Patients experiencing breakthrough pain may be administered a rescue dose, which should be one-fourth to one-half of the current scheduled dose.1-3,13 Patients often take opioid medications regularly for chronic pain associated with prior sickle cell crises; these should also be continued.1
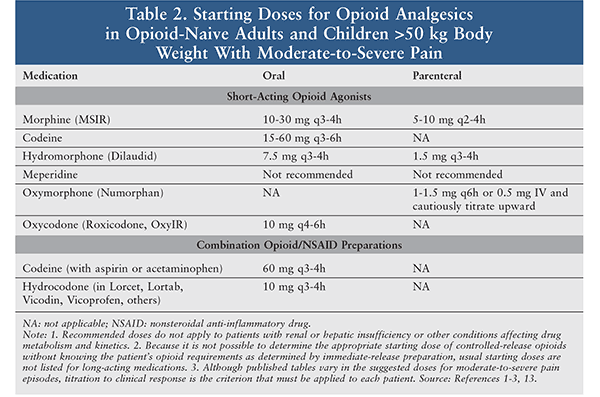
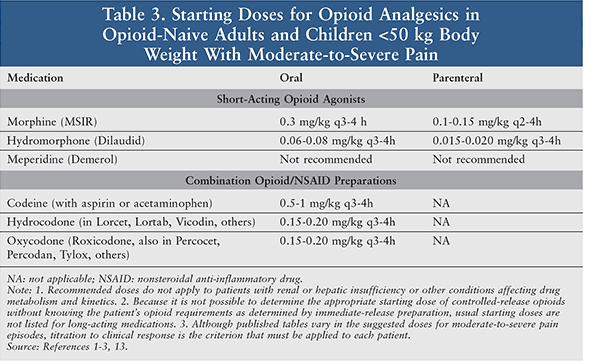
A pain crisis is typically a severe episode of pain, with pain scores >7 out of 10; consequently, pure opioid products are most appropriate as compared with opioid-combination products.1-3,13 The opioid dose varies depending on the severity of the crisis and prior use of opioids. Doses are often selected based upon prior history and current assessment of the clinical situation.1 Patients often require doses higher than the standard analgesic treatment because of pain intensity ,and many patients develop drug tolerance.1-3,13 Severe crises require continuous or intermittent IV administrations on a scheduled basis.1 Due to their hypoxemic state and poor venous access, patients may require oral or SC administration as an alternative.1,13
Upon resolution of acute pain, chronic pain treatment regimens should be reassessed. As the acute pain crisis resolves, providers should taper the patient off the acute opioid regimen to avoid withdrawal syndrome and potential side effects. The management of chronic pain is complex and is further complicated by the level of chronic pain present. The goal is to design a regimen that is effective, with minimal side effects, while balancing concern about drug-seeking behaviors and drug dependence in a population of young patients.1 Patients who use opioids acutely and chronically may experience constipation, sedation, and, rarely, respiratory depression. Additionally, patients with sickle cell disease who take chronic opioids develop physical dependence on the medications and will experience withdrawal symptoms if they have abrupt discontinuation of chronic regimens or large dose reductions without tapering.1,2 Given the current public health threat of opioid overdose in the U.S., the prescribing of naloxone to use if overdose does occur would be appropriate for patients with sickle cell disease who are receiving prescription opioid therapy.
Mild-to-moderate pain episodes can be managed with lower doses of oral opioids at home. Additionally, acetaminophen or a nonsteroidal anti-inflammatory drug (NSAID) such as ibuprofen may be given.11 NSAIDs may also have an additive effect in combination with opioids for severe pain. Hepatic and renal function should be monitored closely because the kidney and liver may be weakened from compensating for hematologic abnormalities. When a patient begins to experience symptoms of breakthrough pain, oral analgesia should be considered for the prevention of a vaso-occlusive crisis. Adjuvant medications may be provided for symptomatic management. Laxatives are often given with opioids to offset constipated side effects and anti-pruritic medications may be prescribed for patients experiencing opioid-induced pruritus.15
Crisis-Reduction Therapy
In 2017, the FDA approved Endari (L-glutamine oral powder) in adult patients and pediatric patients older than age 5 years with sickle cell disease for reducing crisis frequency.15,16 L-glutamine was approved based on data from a randomized, double-blind, controlled trial demonstrating efficacy in reducing pain episodes and acute complications compared with placebo treatment. The study included 230 patients ranging from age 5 to 58 years who had two or more pain crises in the preceding 12-month period. Patients could remain on hydroxyurea therapy and were treated with L-glutamine for up to 12 months. Findings showed a 25% reduction in the frequency of sickle cell crises, a 33% decrease in hospitalization rates, and 60% fewer occurrences of acute chest syndrome in patients administered L-glutamine.12 During the study, patients on L-glutamine experienced a median of three pain crises versus four in the placebo group. The most common adverse effects included constipation, nausea, headache, abdominal pain, cough, extremity and back pain, and back and chest pain. The discontinuation rate for L-glutamine was 2.7%. The recommended dose of L-glutamine is 10 to 30 g per day mixed with 8 oz of room-temperature beverage.13
In 1998, based on the results of the Multicenter Study of Hydroxyurea (MSH), hydroxyurea, a ribonucleotide reductase inhibitor, became the first FDA-approved drug indicated in patients with sickle cell disease for the reduction of frequency in pain episodes, acute chest syndrome hospitalizations, and blood transfusion. Hydroxyurea increases hemoglobin F levels, ultimately inhibiting the polymerization of hemoglobin S within red blood cells.17 The drug also increases mean corpuscular volume, alters the expression of adhesion molecules, and improves cell deformability, which further prevents vaso-occlusion from occurring. A 9-year follow-up of the MSH showed a 40% reduction in mortality in patients who took hydroxyurea compared with those who did not take the medication.2
Patients with sickle cell disease who have three or more pain episodes in a 12-month period or severe pain that impacts their quality of life should be prescribed hydroxyurea 500-mg capsules. The starting dosage for adults is 15 mg/kg/day rounded up to the nearest 500 mg, whereas infants and children should receive 20 mg/kg/day. During follow-up, if hemoglobin F levels are insufficient, the hydroxyurea dosage should be increased by increments of 5 mg/kg/day every 8 weeks up to 35 mg/kg/day until the patient has mild myelosuppression or an absolute neutrophil count of 2,000 to 4,000 cells/mm3. If neutropenia or thrombocytopenia occurs and requires that the dose of hydroxyurea be held, then hydroxyurea should be restarted with a dosage reduction of 5 mg/kg/day in the total daily dose.2,17
The most common adverse effect of the drug is myelosuppression. Therefore, white blood cells, red blood cells, and platelets should be monitored every 2 to 3 months. Other side effects may include ulcers, skin discoloration, or nephrotoxicity. In addition to learning about the chances of having an infant with sickle cell disease, patients of reproductive age should be informed about the drug’s fetal risks; hydroxyurea is still categorized by the old pregnancy classification system as a category D agent. Patients who plan to conceive a child should discontinue the drug prior to becoming pregnant.17,18
Although hydroxyurea has been proven efficacious in randomized, controlled trials, the majority of eligible patients in the U.S. do not take the medication because they fear the side effects and do not believe in the efficacy of the drug.14 Therefore, patients should also be educated on the importance of medication adherence for optimal results.
Follow-up and Monitoring
Patients with a sickle cell disease crisis should follow up with their physician within 1 week of being discharged from the hospital.15 Clinicians should monitor hemoglobin, hematocrit, complete blood count, and reticulocytes to evaluate the patient’s hematologic status. Physicians should also have their patients report their pain score, energy level, and concerning symptoms in order to evaluate home treatment.1-3,19,20 If breakthrough pain that cannot be controlled by oral opioids emerges, the patient needs to immediately return to the hospital for readmission.
For patients on hydroxyurea therapy, white blood cells and hemoglobin F count should be monitored every 3 months to assess for neutropenia and efficacy. Patients on chronic opioid therapy should be asked about pain level, functional status, and adverse effects every 2 to 3 months.1-3,19,20 Depending on the drug’s effectiveness and the patient’s tolerance, dose adjustments may be made.13 Medication adherence is essential for preventing and treating vaso-occlusive episodes. Therefore, patients need to be educated about potential adverse effects, how to take their medication appropriately, and the importance of adhering to the prescribed drugs.
Possibility of a Cure
Hematopoietic stem cell transplants work by replacing the patient’s bone marrow, the source of hemoglobin S production, with blood-forming stem cells from a donor without sickle cell disease. Although bone marrow transplantation can cure sickle cell anemia, it is recommended only for children with frequent refractory pain episodes or severe complications because the procedure is associated with severe side effects and potential mortality.
Gene therapy also offers the possibility of curing the patient of sickle cell disease. Initial studies of gene therapy were noted to have early mortality, which limited the development of the process until more recently. LentiGlobin BB305 is a lenti viral vector engineered by Bluebirdbio to insert a functional human beta-globin gene into the stem cells of patients.21 Recently a case was reported of LentiGlobin use in a boy with a history of numerous vaso-occlusive crises and two episodes of acute chest syndrome who appears to be cured of the blood disorder. At age 13 years, he received an infusion of the antisickling drug LentiGlobin BB305 in Paris, France. More than 15 months after the intervention, the boy has had no occurrences of vaso-occlusive crises, adverse events, or sickle cell disease–related biological measures. Additional data on the LentiGlobin treatment in sickle cell disease is currently being collected in a phase II clinical study in the U.S.22
Future Directions
Research into the management of pain in sickle cell disease continues. Several new targets are under investigation as possible pathways for pain reduction, including P-selectin, histone modulation, demethylating agents, M-TOR effects on hemoglobin F production, and various mast cell targets.23 Numerous agents are being investigated in human and animal models.23
Crizanlizumab, a P-selectin-targeted monoclonal antibody, was studied in 198 patients with sickle cell disease in a phase II double-blind, randomized study.24 The study demonstrated that crizanlizumab increased the median time to first and second pain crises in patients receiving the agent versus placebo; the mean rate of uncomplicated crises was reduced as well. Patients were allowed to be on hydroxyurea therapy while in the study. Adverse effects experienced with this new agent included arthralgia, diarrhea, pruritus, vomiting, and chest pain.24 This new agent has promising results, and further studies are warranted to further evaluate its utility.
Conclusion
Significant morbidity and mortality are associated with sickle cell disease. Fortunately, improvements in survival have occurred. Vaso-occlusive crisis is the most common clinical event in sickle cell disease patients and needs to be managed appropriately by clinicians and family members. During an acute pain episode, providers should assess and monitor patients frequently to achieve pain relief. Patients should be routinely hydrated with fluids and administered IV opioids based on medical history and pain intensity. All sickle cell disease patients with a history of frequently uncontrolled pain episodes should be assessed for hydroxyurea therapy to prevent vaso-occlusive crises. The decision to start hydroxyurea therapy is complicated by patient acceptance, significant toxicities, monitoring and follow-up, and fetal toxicity. L-glutamine is an additional new option to reduce pain crises in patients, with less toxicity than hydroxyurea but more limited patient experience. Continued research is warranted to identify further methods to manage patients with sickle cell disease and to discover potentially curative therapy in the future.
REFERENCES
1. National Institutes of Health. Division of Blood Diseases and Resources. The management of sickle cell disease. NIH publication no. 02-2117, 4th ed. June 2002.
2. Evidence-based management of sickle cell disease: expert panel report, 2014. Pediatrics. 2014;134:e1775.
3. CDC. Sickle cell disease. Data and statistics. www.cdc.gov/ncbddd/sicklecell/data.html. Accessed July 16, 2018.
4. Gardner K, Douiri A, Drasar E. Survival in adults with sickle cell disease in a high-income setting. Blood. 2016;128(10):1436-1438.
5. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762-769.
6. Milner PF, Charache S. Life span of carbamylated red cells in sickle cell anemia. J Clin Invest. 1973;52(12):3161-3171.
7. Baum KF. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987;147(7):1231-1234.
8. Sun PM, Wilburn W, Raynor B, Jamieson D. Sickle cell disease in pregnancy: twenty years of experience at Grady Memorial Hospital, Atlanta, Georgia. Am J Obstet Gynecol. 2001;184(6):1127-1130.
9. Edoh D, Antwi-Bosaiko C, Amuzu D. Fetal hemoglobin during infancy and in sickle cell adults. Afr Health Sci. 2006;6(1):51-54.
10. Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med.1991;325(1):11-16.
11. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Natl Med Assoc. 2010;102(11):1050-1055.
12. Ngo SCA, Bartolucci P, Lobo D, et al. Causes of death in sickle cell disease adult patients: old and new trends. Blood. 2014;124:2715.
13. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematol. 2003;120(5):744-752.
14. Okomo U, Meremikwu MM. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database Syst Rev. 2017;(7):CD005406.
15. Drug Development Technology. Endari for the treatment of sickle cell disease. www.drugdevelopment-technology.com/projects/endari-for-the-treatment-of-sickle-cell-disease. Accessed April 15, 2018.
16. FDA. Drugs. FDA approved L-glutamine powder for the treatment of sickle cell disease. www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm566097.htm. Accessed March 5, 2018.
17. Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low rates of prescription, poor patient compliance, and fears of toxicities and side effects. Expert Rev Hematol. 2010;3(3):255-260.
18. Bridges MDKR. Contraception, pregnancy, and sickle cell disease. Pregnancy and contraception in sickle cell disease. April 4, 2002. http://sickle.bwh.harvard.edu/scd_preg2.html. Accessed February 11, 2018.
19. Anie KA, Grocott, H, White L, et al. Patient self-assessment of hospital pain, mood and health-related quality of life in adults with sickle cell disease. BMJ Open. 2012;2(4):e001274.
20. Hudson S, Wimsatt LA. How to monitor opioid use for your patients with chronic pain. Fam Pract Manag. 2014;21(6):6-11.
21. Bluebirdbio. LentiGlobin. www.bluebirdbio.com. Accessed July 17, 2018.
22. Ribeil J, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Eng J Med. 2017;2:376(9):848-855.
23. Tran H, Gupta M, Gupta K. Targeting novel mechanism of pain in sickle cell disease. Blood. 2017;130(22):2377-2385.
24. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439.
To comment on this article, contact rdavidson@uspharmacist.com.



