US Pharm. 2020;45(5):HS11-HS14.
ABSTRACT: The proposed benefit of pharmacogenetic (PGx) testing in the treatment of major depressive disorder (MDD) is that it facilitates personalized drug selection, thereby minimizing the risk of treatment failure caused by inefficacy or adverse events. Two decades after the mapping of the human genome, however, clinicians continue to be hesitant about PGx testing. Several clinical trials of PGx testing have been conducted, but few have been randomized, controlled, or sufficiently powered to demonstrate a clinically significant advantage for PGx use over treatment-as-usual. PGx testing may be useful for selected patients with inadequately controlled MDD. However, pharmacists should critically evaluate current data before recommending the use of PGx testing in patients with MDD.
Regardless of the research objective, almost every clinical trial investigating major depressive disorder (MDD) delivers the same message: Current treatment-as-usual (TAU) is inadequate.1,2 The pivotal STAR*D trial concluded that 49% of first-episode MDD patients achieved response and 37% achieved remission with first-line antidepressant treatment, but subsequent treatment steps had lower success rates.2 With statements like these, it is unsurprising that psychiatric TAU has garnered the undeserved distinction of being trial-and-error, often with an emphasis on “error.”3
History of Pharmacogenetics
Companies have capitalized on psychiatry’s trial-and-error reputation by developing and marketing pharmacogenetic (PGx) testing products that they claim can provide prescribers with the necessary information to choose a genetically compatible medication of optimal therapeutic dosage at the start, thereby decreasing the risk of adverse drug events (ADEs) and maximizing treatment success.4
The human genome was mapped in 2001, and PGx was expected to revolutionize clinical practice by driving personalized PGx-driven drug selection.5,6 Two decades later, however, clinicians continue to be hesitant about PGx testing, most commonly citing concerns about methodological flaws in efficacy trials.1,7 This article will provide an overview of the concepts behind PGx testing and critically evaluate recently published evidence to determine the clinical utility of PGx testing in patients with MDD.
The idea that genetics determines how the body interacts with substances is quite old. In the sixth century bc, the Greek scientist Pythagoras made the first PGx observation when he realized that some groups of people became ill after eating fava beans, but others did not. (Incidentally, the illness he witnessed was hemolytic anemia caused by a genetically inherited defect in the G6PD enzyme.)8
PGx Basics
PGx centers on the simple pharmacokinetic premise that there is a correlation between a drug’s concentration and its effect; therefore, it is assumed that any pathway, mechanism, or factor that changes a drug’s concentration will also play a role in that drug’s effect.5,9 PGx and pharmacokinetics intersect at drug metabolism. Metabolism is largely controlled by CYP450 enzymes, which, because of their multiallelic structure, are prone to genetic variations (polymorphisms).6 PGx-testing companies use genotyping technology to isolate phenotypes for various genes.4 Depending on the polymorphism present, the phenotype is categorized as one of following: poor metabolizer (PM), intermediate metabolizer (IM), extensive metabolizer, or ultrarapid metabolizer.8 As TABLE 1 denotes, each phenotype has a different level of metabolic activity, so knowing a patient’s phenotypical profile theoretically could assist drug selection and dosing.10 However, genotyping can measure only the effect of genetic polymorphisms on CYP enzymes. Because CYP expression is also influenced by environmental (other medications, certain foods, smoking) and physiological factors (obesity, hormones, pregnancy, hepatic function), the PGx test may only partially explain a patient’s response to a drug.5 One clinically relevant example is the emergence of new side effects when a patient with a CYP2D6 IM phenotype begins taking a CYP2D6 inhibitor.5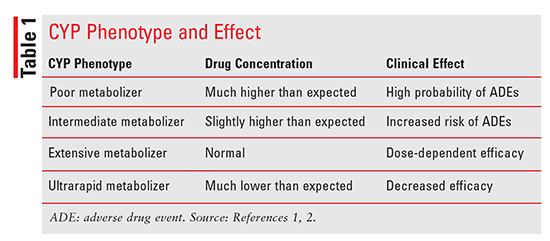
Clinical Controversy
The confounding effects of environmental and physiological factors on clinical response can be overcome only with strong clinical-trial design.1 Clinical trials that are adequately powered with representative population samples and are randomized, controlled, prospective, and blinded are necessary to differentiate the clinical utility of PGx testing from a false-positive result.1,4 However, the FDA does not regulate the laboratory-testing market. Laboratory-testing companies are not required to adhere to the FDA’s rigorous standards for clinical-trial design.4
As of 2018, there were 10 studies evaluating PGx for MDD. No test was fully blinded or had a suitable control group.1 Trials that assess the efficacy of PGx most often use TAU as the control group. To ensure that it is an adequate comparator, TAU should be standardized and protocol-based (i.e., STAR*D, Texas Medication Algorithm Project).1 Otherwise, results showing superiority of PGx testing to TAU could actually be due to ineffective or intolerable drugs used as TAU. To date, no clinical trial has used a protocol-based comparison.1
Further, industry funding notwithstanding, each company’s PGx test is proprietary and varies according to the genes tested and the clinical information reported. Trial results using a specific PGx test cannot be compared or extrapolated to another company’s test.10
Current Evidence
The two PGx trials discussed here are adequately powered to detect clinically meaningful outcomes. They are prospective, randomized, and controlled with partial blinding of patients and raters. Although they are not methodologically flawless, they represent the strongest clinical evidence available.1
GUIDED Trial: The GUIDED trial, the largest PGx study to date, involved 60 sites and enrolled 1,167 subjects with moderate-to-severe MDD and an inadequate response to current treatment.11 Subjects were randomized 1:1 to either PGx-guided treatment using the GeneSight test or TAU and were followed for 24 weeks. Per protocol, subjects and raters in both arms were unblinded by week 12, but only the data from baseline to week 8 (the study endpoint) were considered blinded. Clinicians were unblinded throughout the trial.11
The primary outcome was symptom improvement at week 8, measured by percent change in Hamilton Rating Scale for Depression (HAM-D) scores from baseline. Secondary outcomes were response and remission rates at week 8. Response was defined as a 50% or greater decrease in HAM-D score from baseline to study endpoint; remission was a score of 7 or less at study endpoint.11 Treatment groups were balanced and had similar baseline demographics, disease severity, and metabolizer phenotypes. The mean number of previously failed medications was 3.51 for both groups.11
Differences in the primary outcome of symptom improvement were not significant despite being adequately powered, nor were there any differences in the number of ADEs experienced by each group at endpoint. However, there was a statistically higher response and remission at week 8 (TABLE 2) in the PGx group versus the control group (response: 26% vs. 19.9%, P = .013; remission: 15.3% vs. 10.1%, P = .007).11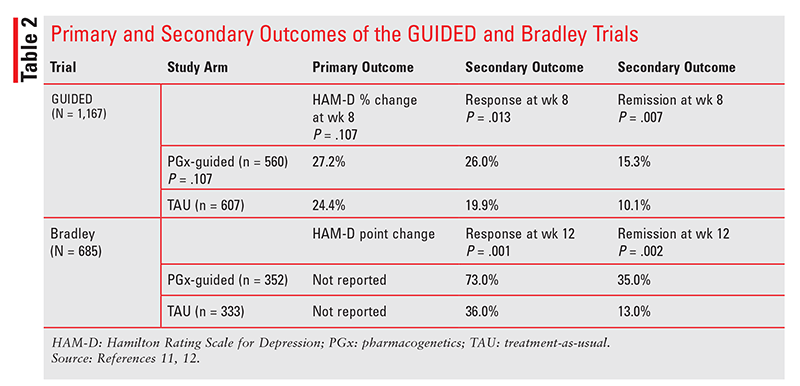
The researchers claimed that the ability to switch subjects from genetically incompatible medications to medications that are compatible is a benefit of using PGx testing, as switching could increase treatment success.11 In this study, medications were considered congruent if they were in the “use as directed” or “use with caution” categories in the PGx report. Incongruent medications were in the “use with increased caution and with more frequent monitoring” category. However, at baseline, 80% of PGx-guided subjects and 77.5% of controls were already taking congruent medications.11 By week 8, 91% of PGx-guided subjects were congruent and control subjects remained unchanged at 77.5%. When only subjects taking incongruent medications at baseline (n = 213) were assessed, significant increases in response and remission were found in the 77 subjects who were switched to a congruent medication compared with the 136 subjects who remained on an incongruent one until week 8 (response, 28.5% vs. 16.7%, P = .04; remission, 21.5% vs. 8.5%, P = .007).11 However, since three-fourths of subjects were already in line with PGx recommendations, the clinical utility of PGx testing was questionable.1,11
As shown in TABLE 3, the use of selective serotonin reuptake inhibitors (SSRIs) steadily decreased from baseline to week 8, whereas serotonin-norepinephrine reuptake inhibitor (SNRI) use increased.11 Switching to an SNRI after SSRI treatment failure is the standard of care in MDD guidelines, even without the use of PGx testing.2 The modest positive results should be weighed against the possibility that the increased treatment effect may be largely due to the switch to SNRIs.1,2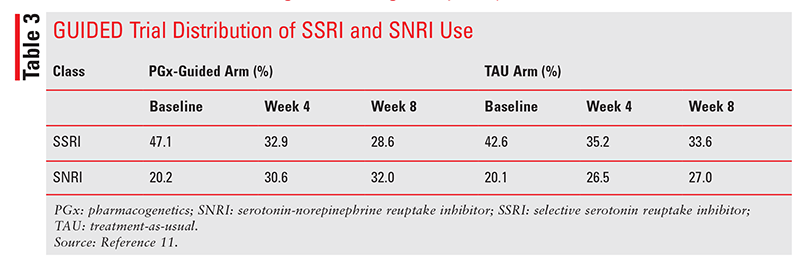
Bradley Trial: This trial involving 20 clinical sites included 685 subjects with MDD or anxiety who were either treatment-naïve or inadequately controlled.12 Subjects were randomized 1:1 to either PGx-guided treatment using the NeuroIDgenetix test or TAU and were prospectively followed for 12 weeks. Subjects and raters remained blinded until the end of the trial. Clinicians were unblinded to PGx results, but all assessments were made by raters.12
The primary efficacy endpoint was change in HAM-D score in subjects with depression from baseline to 4-, 8-, and 12-week follow-up visits. Only subjects with moderate-to-severe HAM-D scores (8 or higher) at baseline were included in the efficacy analysis. Treatment groups were similar and balanced with regard to severity and metabolizer phenotype.12
Primary efficacy endpoint data were not disclosed; however, compared with controls, PGx subjects with severe MDD experienced significantly higher rates of remission at week 12 (35% vs. 13%; P = .002; odds ratio [OR], 3.54; number needed to treat [NNT], 4.6) and of response (73% vs. 36%; P = .001; OR, 4.72; NNT, 2.7) (TABLE 2). ADEs were similar in number and severity between groups.12 The number of medication changes (added or discontinued medication) in the PGx group was significantly higher than in the control group (n = 552 vs. n = 442; P <.0001), but the number of dose changes was similar.12 Notably, medication changes in the PGx group were congruent with PGx test results 70% of the time, compared with 29% (due to chance) in the control group. Reasons that clinicians chose an incongruent medication were not given.12
Since the percentage of incongruent versus congruent medications at baseline and the specific medications taken were not specified, the extent to which PGx-guided care contributed to the positive outcome, as opposed to a non–PGx-guided change to a more efficacious medication, is unknown.1,4
Analysis: According to the FDA, 13 antidepressants have sufficient PGx evidence to warrant changes to therapy, depending on the phenotype.13 These actionable drug/gene pairs are as follows: CYP2D6 PM—amoxapine, clomipramine, desipramine, doxepin, fluvoxamine, imipramine, protriptyline, venlafaxine, and vortioxetine; CYP2C19 PM—citalopram, doxepin, and escitalopram.13,14 All of them are metabolized by CYP2D6 or CYP2C19.13 Prevalence studies estimate that 5% to 10% of U.S. patients are 2D6 PMs and 2% to 15% are 2C19 PMs.14 In the GUIDED trial, 28.9% and 6.7% of subjects were 2D6 or 2C19 PMs, respectively; the Bradley trial included 9.7% and 6%.11,12 Therefore, questions should be raised regarding the external validity of the GUIDED trial because its 2D6 PM population, which would require the most medication changes, is much larger than would be encountered in clinical practice. Further, since such a small population would be expected to have the phenotypes that would necessitate any medication changes, it may not be cost-effective to suggest preemptive PGx testing for all patients with MDD.4
The Pharmacist’s Role
With formal training in pharmacokinetics and pharmacology, pharmacists can play a prominent role in the implementation of PGx testing.10 In 2015, the American Society of Health-Systems Pharmacists published a position statement endorsing pharmacist involvement in all stages of PGx testing, including patient selection, posttest patient counseling, and clinical consultation with prescribers.3 Until data suggest a definitive benefit to PGx testing, pharmacists should continue to recommend optimizing evidence-based treatment strategies prior to suggesting PGx testing for patients with MDD.1,4,10,15
Conclusion
At this time, preemptive PGx testing is not recommended.4 However, for patients with inadequately controlled MDD who have failed previous trials of guideline-based pharmacotherapy (at an adequate dose for adequate duration) and patients who experience intolerable ADEs from antidepressants at low doses, PGx testing may be useful.10 If switching to a genetically congruent medication is supported by clinical judgment, the pharmacist should work with the prescriber to create a cross-taper schedule in order to avoid discontinuation syndrome. Ideally, the information gathered from a PGx test will serve a supportive, not prescriptive, role.8
REFERENCES
1. Zubenko GS, Sommer BR, Cohen BM. On the marketing and use of pharmacogenetic tests for psychiatric treatment. JAMA Psychiatry. 2018;75:769-770.
2. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905-1917.
3. Brown L, Eum S, Haga SB, et al. Clinical utilization of pharmacogenetics in psychiatry—perspectives of pharmacists, genetic counselors, implementation science, clinicians, and industry. Pharmacopsychiatry. 2019 Sep 11 [Epub ahead of print].
4. de Leon J, Spina E. What is needed to incorporate clinical pharmacogenetic tests into the practice of psychopharmacotherapy? Expert Rev Clin Pharmacol. 2016;9:351-354.
5. Baskys A. Application of pharmacogenetics in clinical practice: problems and solutions. J Neural Transm (Vienna). 2019;126:109-113.
6. Ravyn D, Ravyn V, Lowney R, Nasrallah HA. CYP450 pharmacogenetic treatment strategies for antipsychotics: a review of the evidence. Schizophr Res. 2013;149:1-14.
7. Muflih S, Bleidt BA, Lafferty L, et al. Measuring knowledge and attitudes towards the utilization of pharmacogenetic testing among physicians. J Pharm Health Serv Res. 2019;10:227-234.
8. Müller DJ, Rizhanovsky Z. From the origins of pharmacogenetics to first applications in psychiatry. Pharmacopsychiatry. 2019 Sep 23 [Epub ahead of print].
9. Spina E, Hiemke C, de Leon J. Assessing drug-drug interactions through therapeutic drug monitoring when administering oral second-generation antipsychotics. Expert Opin Drug Metabol Toxicol. 2016;12:407-422.
10. de Leon J. The future (or lack of future) of personalized prescription in psychiatry. Pharmacol Res. 2009;59:81-89.
11. Greden JF, Parikh SV, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 2019;111:59-67.
12. Bradley P, Shiekh M, Mehra V, et al. Improved efficacy with targeted pharmacogenetic-guided treatment of patients with depression and anxiety: a randomized clinical trial demonstrating clinical utility. J Psychiatr Res. 2018;96:100-107.
13. Hicks JK, Bishop JR, Gammal RS, et al. A call for clear and consistent communications regarding the role of pharmacogenetics in antidepressant pharmacotherapy. Clin Pharmacol Ther. 2020;107:50-52.
14. Hicks JK, Bishop JR, Sangkuhl K. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98:127-134.
15. de Leon J. Have we successfully implemented CYP2D6 genotyping in psychiatry? Expert Opin Drug Metabol Toxicol. 2017;13:1201-1203.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



