Clinical literature has recently reported that gout is the most common inflammatory arthritis in the United States, with 3 to 5 million sufferers.1,2 Both the incidence and the prevalence of gout appear to be increasing worldwide.3 Gout is perhaps the oldest known type of arthritis; it has been colorfully depicted in art and literature along with commentaries on the moral character of the gout sufferer (FIGURE 1). Literature accounts have referred to gout’s association with rich foods and excessive alcohol consumption—thus the description, “the disease of kings.”
Gout is a monosodium urate, monohydrate crystal deposit disease with a very rich history mirroring the evolution of medicine itself.4,5 It was among the earliest diseases to be recognized as a clinical entity. Since gout has been recognized for so many centuries, its diagnosis and treatment generally have not elicited much interest; thus, the management of gout is a challenge for the clinician caring for the patient with this disease.6
Recent medical literature recognizes that most patients with gout visit a primary care physician for disease management, but there are challenges to diagnosing and treating gout in this setting.7 Further, Weaver et al stated that the arrival of newer investigational agents in the market has prompted rheumatologists to consider how they can share current information to improve gout management.7 It is this concept of sharing current information on the management of gout that is the main impetus for the preparation of this review. It is hoped that pharmacists will be empowered with this knowledge to assist the prescribing clinician to maximize patient outcomes when treating gout. First, to serve as a foundation, new insights into the pathogenesis of hyperuricemia and gout will be discussed. Second, risk factors, typical presentation of symptoms, and key diagnostic parameters will be reviewed so that the pharmacist may achieve an appreciation of the disease. Finally, nonpharmacologic treatment modalities and both current as well as newer investigational therapeutics will be offered so that the pharmacist may facilitate greater patient adherence through medication counseling.
Pathogenesis
Biologically significant hyperuricemia occurs when serum urate levels exceed solubility (~6.8 mg/dL). Hyperuricemia is a common serum abnormality that does not always progress to gout. Humans generate about 250 to 750 mg of uric acid per day. The uric acid comes from dietary purines and the breakdown of dying tissues. The exact cause of gout is not yet known, although it may be linked to a genetic defect in purine metabolism. Uric acid, the most insoluble of the purine substances, is a trioxypurine containing three oxygen groups. The pathogenesis of gout starts with the crystallization of urate within the joint, bursa, or tendon sheath, which leads to inflammation as a result of phagocytosis of monosodium urate crystals; the disease is usually associated with an elevated concentration of uric acid in the blood.2,8 Specifically, uric acid is a breakdown product of the purines adenine, guanine, hypoxanthine, and xanthine. Adenine and guanine are found in both DNA and RNA. Hypoxanthine and xanthine are not incorporated into the nucleic acids as they are being synthesized, but they are important intermediates in the synthesis and degradation of the purine nucleotides. Both undissociated uric acid and monosodium salt, which is the primary form found in the blood, are only sparingly soluble.
The amount of urate in the body depends on the balance between dietary intake, synthesis, and excretion.9 In people with primary gout, defects in purine metabolism lead to hyperuricemia, or high levels of uric acid in the blood. This can be caused by increased production of uric acid, abnormal retention of uric acid, or both. Urate in the blood can accumulate either through an overproduction or an underexcretion of uric acid. Hyperuricemia results from the overproduction of urate found in 10% of gout patients and from underexcretion of urate found in the remaining 90%.9 The majority of patients with endogenous overproduction of urate have the condition as a result of salvaged purines arising from increased cell turnover in proliferation and inflammatory disorders, from pharmacologic intervention resulting in increased urate production, and from tissue hypoxia.9
The renal mechanism for handling urate is one of glomerular filtration followed by partial tubular reabsorption.10 The final fractional excretion of uric acid is about 20% of what was originally filtered. Uric acid levels independently predict renal failure in patients with preexisting renal disease. Hyperuricemia causes interstitial and glomerular changes that are independent of the presence of crystal, and the changes very much resemble what hypertensive changes would look like chronically. In addition, serum hyperuricemia is epidemiologically linked to hypertension and seems to be an independent factor for the development of hypertension. Finally, hyperuricemia is defined as a serum uric acid level greater than 6.8 mg/dL. Serum uric acid can be normal, especially during the gout attack. The target goal for uric acid treatment is to achieve a level less than 6.0 mg/dL.
Risk Factors for Gout
A number of references by Choi et al have identified, explained, and reviewed the risk factors for the development of gout.11-13 Nonmodifiable risk factors include being a male or a postmenopausal female, genetic influences, end-stage renal disease, and resulting major organ transplantation. Its prevalence increases with age, from 1.8/1,000 in people under the age of 45 years to 30.8/1,000 in those over age 65.8 Elevated serum urate levels are also associated with increased risk.8 Hypertension is a definite risk factor, as a significant percentage of patients with hyperuricemia will develop hypertension. Hyperuricemia and gout have been linked to other disease states including metabolic syndrome, cardiac disease, stroke, and renal disease.8 The risk of gout correlates with truncal obesity, as measured by body mass index and waist-to-hip ratios.8,11
Avoidable risk factors include diet and medications. Foods that have been implicated in causing gout are red-organ meats, seafood, and foods containing high-fructose corn syrup. Fructose has been recognized as a cause of hyperuricemia.8,14-16 Choi et al conducted a small prospective study that investigated the ability of diets high in fructose to induce higher serum urate levels relative to diets high in glucose or low in carbonates.16 High alcohol intake, especially beer, is also a risk factor. The presence of guanosine in beer has been identified as the cause of gouty attacks.
Certain drugs used to treat gout, particularly thiazide diuretics and the cyclosporine administered to transplant patients, have been implicated with gouty attacks. Despite the cardioprotection offered by low-dose 81-mg aspirin, this drug may be associated with the precipitation of gout.8,17 Commonly, the use of cyclosporine has been reported to cause a rapidly occurring type of gout, swiftly ascending and polyarticular in many cases. Roubenoff validates that these risk factors are increasing by reporting that gout incidence and prevalence have increased by twofold from 1970 to 1990.18 Furthermore, Wallace et al have reported that the prevalence of gout has increased by two cases per 1,000 patients during the 1990s because of lifestyle changes.19
Typical Presentation of Gout
Gouty attacks are usually associated with a precipitating event.6,20 These attacks consist of intense pain involving the lower extremity, with 80% of first attacks involving a monoarticular joint; however, after long periods of time, gout attacks may become polyarticular.6,20,21 This pain and inflammation is a result of a dramatic inflammatory response. Some authors have estimated that between 50% to 90% of the initial attacks occur in the first metatarsophalangeal joint (podagra).6,21-23 In postmenopausal women, the distal interphalangeal joints may be involved.6 Attacks often occur at night and are associated with a precipitating event.6,20 Acute gouty arthritis may be accompanied by low-grade fever, chills, and malaise.6,21,23 The majority of patients experience a second acute gout attack within 1 year of the first episode.24 Untreated initial acute gout attacks resolve completely within 3 to 14 days.6,20,23
There are four clinical stages of gout.23 At serum urate concentrations greater than 6.8 mg/dL, urate crystals may start to deposit. Hence, the first stage of gout is known as asymptomatic hyperuricemia. During this first period, urate deposits may directly contribute to organ damage. After sufficient urate deposits have developed around a joint and some traumatic event triggers the release of crystals into the joint space, a patient will suffer an acute gout attack and move into the second stage, known as acute gouty arthritis. During this second stage, acute inflammation in the joint caused by urate crystallization and crystal phagocytosis is present. This episode is known as a “flare” and is self-resolving and likely to recur. The interval between acute flare gout attacks with persistent crystals in the joints is the third stage and is known as an intercritical period. When crystal deposits continue to accumulate, patients develop chronically stiff and swollen joints leading to the final stage—advanced gout, which includes the long-term complications of uncontrolled hyperuricemia characterized by chronic arthritis and tophi. The nodular mass of uric acid crystals is described as a tophus and is characteristically deposited in different soft tissue areas of the body in gout. This advanced stage of gout is uncommon because it is avoidable with interventional therapy.23
Diagnosis
The diagnosis of gout can be straightforward. The only way to establish the diagnosis with certainty is to demonstrate uric acid crystals in synovial fluid or tophi.7 Polarizing microscopic examination of synovial fluid reveals negatively birefringent crystals, confirming the diagnosis of gout. It must be recognized that normal uric acid levels are observed in approximately 50% of acute gouty flares.7
Dalbeth and McQueen’s review summarizes recent advances in plain radiography and advanced imaging for gout, calcium pyrophosphate dihydrate crystal arthropathy, and basic calcium phosphate crystal arthropathy.24 They suggest that high-resolution ultrasonography may improve noninvasive diagnosis of the crystal-induced arthropathies and allow for monitoring of intra-articular tophi. They also determined that computed tomography provides excellent definition of tophi and bone erosion, and three-dimensional computed tomography assessment of tophus volume is a promising outcome measure in gout.24 Finally, they state that magnetic resonance imaging is also a reliable method for assessment of tophus size in gout and has an important role in detection of complications of the disease in clinical practice.

Treatment of Gout
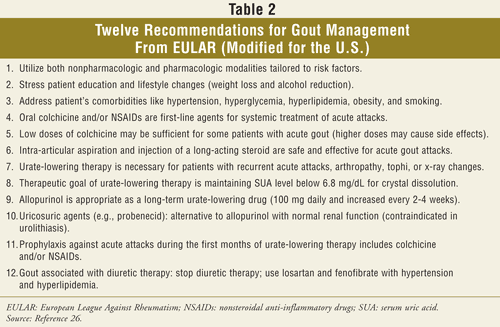
Key elements necessary to improve clinical outcomes in gout management include enhancing health professional and patient education as well as exploring novel urate-lowering agents. One of the most valuable health care professionals when assisting clinicians in the treatment of gout is the pharmacist. Pharmacists can appreciate that the optimal treatment for gout requires both adjunctive nonpharmacologic as well as pharmacologic interventional therapies (TABLE 1). A practicing pharmacist is directed to read the Becker and Chohan editorial, which suggests that successful gout management is possible by embracing the 12 evidence-based recommendations from the European League Against Rheumatism (EULAR) (TABLE 2).25,26 Treatment and prevention of acute gout flares, as well as the management of hyperuricemia and gout, can best be appreciated by patients with a brief narrative and be accentuated graphically through easy-to-read tables that the pharmacist may access quickly when a question arises.
A treatment regimen must be individually tailored to each patient. The treatment of gout has three main components: therapy of the acute attack, prophylaxis against gout flares, and management of hyperuricemia.8 Several aspects must be independently considered when planning to treat a patient with gout. Given that gout is a reversible urate crystal deposit disease, the main objective is to eliminate the urate crystals from the joints and other structures.27 Li-Yu et al determined through a 10-year prospective investigation that serum urate levels should be reduced to below 6.0 mg/dL in order to eliminate crystals.28
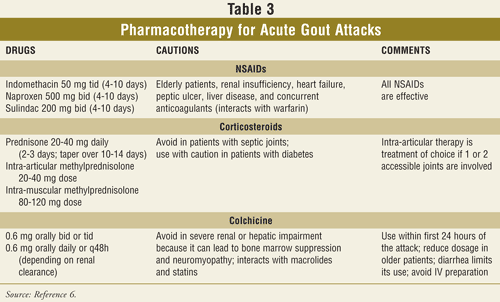
Pharmacotherapy for Acute Gout Attacks: Medications used to treat an acute gout attack include nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, and corticosteroids. A combination of these may also be necessary. A summary of the pharmacologic agents used to treat acute gout is shown in TABLE 3.6-8,25-27 These medications have no effect on the serum uric acid level. The classic antidote for gouty arthritis is colchicine. The most common associated adverse drug event reported with colchicine use is diarrhea.6 However, even low-dose colchicine may be associated with severe adverse effects and toxicity such as myopathy and myelosuppression.6,8,27 Monitoring of serum troponin levels during an acute colchicine overdose may help avoid vascular collapse.29
Guidelines indicate that fast-acting oral NSAIDs should be used during acute attacks if there are no contraindications.6-8,26 Since there are no significant clinical differences among NSAIDs, the choice of agent should be based on the agent’s side-effect profile, it’s cost, and patient’s ability to adhere to the prescribed agent.6,8,25,27 Suppressive therapy to prevent flares usually involves colchicine or NSAIDs.8 An important factor in choosing therapeutic agents for an acute attack is the presence of comorbidities. The most common therapy for acute gout in the setting of acute or chronic renal or hepatic failure is corticosteroids.8 If NSAIDs and colchicine are contraindicated because of patient comorbidities, intra-articular aspiration and injection of a corticosteroid is an effective treatment of an acute attack of gout once the possibility of a septic joint has been eliminated.6
Urate-Lowering Therapy: The therapeutic goal of urate-lowering therapy is to promote dissolution of the urate crystals and to prevent crystal formation.6,28 In addition, urate-lowering therapy is used to prevent disease progression, reduce the frequency of acute attacks, and maintain and improve quality of life. Treatments for chronic gout are aimed at reducing serum urate levels to less than 6.0 mg/dL in order to dissolve existing crystals and prevent formation of new ones.8 Dore recommends that patients who overproduce urate should be treated with allopurinol, as this drug has the advantage of being effective for both overproducers and underexcretors.6 Patients who underexcrete urate despite near-normal creatinine clearance levels should be treated with uricosurics. Urate-lowering therapy should be lifelong. If an acute flare occurs when urate-lowering therapy is initiated, therapy should not be discontinued, because doing so will result in fluctuating urate levels.30 Initiating urate-lowering therapy can mobilize urate deposits, which may precipitate an attack because of rapid serum uric acid lowering.7 The practice of using concomitant gastroprotectant NSAID and colchicine prophylaxis with the initialization of urate-lowering therapy has been suggested.7,25
Pharmacists can be a tremendous resource by informing the clinician about potential drug interactions and side effects of urate-lowering agents to maximize therapeutic outcomes. Finally, pharmacists must remember that treatment of asymptomatic hyperuricemia is not recommended.7
Since 1965, one traditional approach to the treatment of gout has been the drug allopurinol, an isomer of hypoxanthine. Allopurinol is a substrate for xanthine oxidase. The product binds so tightly that the enzyme is now unable to be oxidized in its normal substrate. Uric acid production is diminished, and xanthine and hypoxanthine levels in the blood rise. These are more soluble than urate and are less likely to deposit as crystals in the joints. The allopurinol dose must be adjusted in patients with renal impairment.6 Allopurinol is often started at 100 mg per day, and the daily dosage is increased in 100 mg increments every 2 to 4 weeks.8,25,26 The usual dosage range for allopurinol is 200 to 300 mg/day for mild gout and 400 to 600 mg/day for cases of moderate and severe gout. Up to 5% of patients are unable to tolerate allopurinol due to adverse effects such as rash, nausea, and bone marrow suppression.31 If a severe rash occurs, the pharmacist should advise discontinuation of allopurinol. Allopurinol has fewer drug interactions than uricosuric agents.6 Despite allopurinol’s limitations, it is used extensively for most gouty patients and is considered safe and effective.27
Emerging Therapies: Uricosurics are considered second-line therapy for patients who are intolerant to allopurinol. Of all the older urate-lowering drugs, probenecid or sulfinpyrazone may be used in patients refractory to allopurinol therapy.27 In the U.S., probenecid is the only potent uricosuric agent available.8 Probenecid is most useful in patients with mild gout and normal renal function. Its mechanism of action is inhibition of the uric acid transporter (URAT1) involved in the reabsorption of uric acid.8 Uricosuric therapy is contraindicated in patients with a history of nephrolithiasis and is not effective in patients with a creatinine clearance of less than 50 mL/min. Finally, both losartan and fenofibrate have slight uricosuric properties and may be useful as adjunctive therapy in gout patients with comorbidities of hypertension and hyperlipidemia.6,32
Febuxostat is a potent, new selective xanthine oxidase inhibitor that received FDA approval in February 2009 for the management of hyperuricemia in patients with gout.8,33,34 This agent is not a purine analog and has a mechanism similar to that of allopurinol. The recommended starting dose of febuxostat is 40 mg once a day. For patients who do not achieve a serum uric acid less than 6 mg/dL after 2 weeks with 40 mg, increasing the dose to 80 mg is recommended.34 Febuxostat has demonstrated efficacy superior to allopurinol.27,34 It is primarily metabolized by the liver and may be an alternative agent for patients with renal insufficiency. The adverse-effect profile for febuxostat includes elevation in liver enzymes, rash, diarrhea, and headache.8 The manufacturer has reported that febuxostat has a higher rate of risk of cardiovascular thromboembolic events compared to allopurinol.34 Finally, febuxostat is contraindicated in patients treated with azathioprine, mercaptopurine, or theophylline.34
Uric acid oxidase, also known as uricase, is an enzyme that catalyzes the conversion of uric acid to allantoin and is present in all mammals except humans and higher primates.27 There is interest in using uricase therapies to lower serum uric acid. Rasburicase, a recombinant uricase IV product indicated for tumor lysis syndrome, might be successfully used in unusually severe cases of gout.35 Rasburicase has a black box warning for anaphylaxis, hemolysis, and methemoglobin. Pegloticase (pegylated recombinant porcine uricase) has also shown urate-lowering efficacy.14,36 Adding polyethylene glycol (PEG) prolongs the half-life of uricase and decreases the antigenicity. Intravenous administration of PEG-uricase has been investigated for the potential treatment of severe tophaceous gout in patients who are hypersensitive to allopurinol.37
Pharmacists should appreciate the relative contraindications to both NSAIDs and corticosteroids as symptomatic therapies. Therefore, attention has been directed to the recent advances in the understanding of gouty inflammation and the proinflammatory role of several cytokines in the pathophysiology of acute gout.25,38 Early small clinical trials have identified interleukin-1B as the most prominent in acute gout. The practice of inhibiting interleukin-1 may be efficient and safe in terminating the symptoms of acute gouty arthritis.25
Conclusion
Gout is a monosodium urate, monohydrate crystal deposit disease. It was among the earliest diseases to be recognized as a clinical entity. Clinical pharmacists need to be empowered with knowledge to assist prescribing clinicians in order to maximize therapeutic outcomes when treating gout. To achieve this goal, a foundation of new insights into the pathogenesis of hyperuricemia and gout has been reviewed. Risk factors, typical presentation of symptoms, and key diagnostic parameters have been offered so that pharmacists can achieve an appreciation of gout as a significant disease. Both nonpharmacologic modalities and pharmacologic therapies have been discussed so that greater patient adherence through medication counseling can be achieved.
REFERENCES
2. Lawrence RC, Felson DT, Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26-35.
3. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75(suppl 5):S9-S12.
4. Pillinger MH, Rosenthal P, Abeles AM. Hyperuricemia and gout. Bull NYU Hosp Jt Dis.
5. Nuki G, Simkin PA. A concise history of gout and hyperuricemia and their treatment. Arthritis Res Ther. 2006;8(suppl 1):S1.
6. Dore RK. Gout: what primary care physicians want to know. J Clin Rheumatol. 2008;14(suppl 5):S47-S54.
7. Weaver AL, Cheh MA, Kennison RH. How PCP education can impact gout management: the gout essentials. J Clin Rheumatol. 2008;14(suppl):S42-S46.
8. Mandel DA, Simkin PA. Gout: update on pathogenesis, diagnosis, and treatment. New Develop Rheum Dis. 2007;20-25. www.medpub.com/NDRDv4n3_ 2007;65:215-221.
9. Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499-516.
10. Wyngaarden JB. The role of the kidney in pathogenesis and treatment of gout. Arthritis Rheum. 1958;1:191-203.
11. Choi HK, Atkinson K, Karlson EW, et al. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow up study. Arch Intern Med. 2005;165:742-748.
12. Choi HK, Atkinson K, Karlson EW, et al. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363:1277-1281.
13. Choi HK, Atkinson K, Karlson EW, et al. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350:1093-1103.
14. Pillinger MH, Keenan RT. Update on the management of hyperuricemia and gout. Bull NYU Hosp Jt Dis. 2008;66:231-239.
15. Emmerson BT. Effect of oral fructose on urate production. Ann Rheum Dis. 1974;33:276-280.
16. Choi JW, Ford ES, Gao X, Choi HK. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008;59:109-116.
17. Schlesinger N, Schumacher HR. Update on gout. Arthritis Rheum. 2002;47:563-565.
18. Roubenoff R. Gout and hyperuricemia. Rheum Dis Clin North Am. 1990;16:539-550.
19. Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31:1582-1587.
20. Hawkins DW, Rahn DW. Gout and hyperuricemia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 5th ed. New York, NY: McGraw-Hill; 2002:1659-1664.
21. Becker MA, Jolly M. Clinical gout and the pathogenesis of hyperuricemia. In: Koopman WJ, Moreland LW, eds. Arthritis and Allied Conditions. 15th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:2303-2339.
22. Worthmann PL, Kelly WN. Gout and hyperuricemia. In: Harris ED Jr, Budd RC, Genovese MC, et al, eds. Kelly’s Textbook of Rheumatology. 7th ed. Philadelphia, PA: WB Saunders Company; 2005:1402-1429.
23. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75(suppl 5):S5-S8.
24. Dalbeth N, McQueen FM. Use of imaging to evaluate gout and other crystal deposition disorders. Curr Opin Rheumatol. 2009;21:124-131.
25. Becker MA, Chohan S. We can make gout management more successful now. Curr Opin Rheumatol. 2008;20:167-172.
26. Zhang W, Doherty M, Bardin T, et al. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1312-1324.
27. Pascual E, Sivera F. Therapeutic advances in gout. Curr Opin Rheumatol. 2007;19:122-127.
28. Li-Yu J, Clayburne G, Sieck M, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol. 2001;28:577-580.
29. van Heyningen C, Watson ID. Troponin for prediction of cardiovascular collapse in acute colchicine overdose. Emerg Med J. 2005;22:599-600.
30. Schumacher HR Jr, Chen LX. The practical management of gout. Cleve Clin J Med. 2008;75(suppl 5):S22-S25.
31. Schlesinger N. Management of acute and chronic gouty arthritis: present state-of-the-art. Drugs. 2004;64:2399-2416.
32. Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
33. Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
34. Uloric (febuxostat) package insert. Deerfield, IL: Takeda Pharmaceuticals North America; February 2009. www.uloric.com. Accessed April 13, 2009.
35. Vogt B. Urate oxidase (rasburicase) for treatment of severe tophaceous gout. Nephrol Dial Transplant. 2005;20:431-433.
36. Biggers K, Scheinfeld N. Pegloticase, a polyethylene glycol conjugate of uricase for the potential intravenous treatment of gout. Curr Opin Investig Drugs. 2008;9:422-442.
37. Terkeltaub R, Bushinsky DA, Becker MA. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res Ther. 2006;8(suppl 1):S4.
38. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
To comment on this article, contact rdavidson@jobson.com.


