US Pharm. 2010;35(8):44-56.
Over the past several years, the incidence of invasive fungal infections has dramatically increased.1,2 In fact, Candida species (spp) are the fourth most common cause of nosocomial bloodstream infections in the United States.3 Other fungal pathogens such as Aspergillus spp, zygomycetes, Fusarium spp, and Scedosporium spp have become more common at causing invasive infections. There are multiple reasons for this increased incidence, including the use of broad-spectrum antibiotics, central venous catheters, and prosthetic devices.4-6 Additionally, patients with burns and neutropenia or those who are the recipients of parenteral nutrition, renal replacement therapy, immunosuppressive therapy, and antineoplastic agents can also be predisposed to fungal infections.4-6 Invasive fungal infections can have a significant impact on patient morbidity and mortality.
Fungal infections can be difficult to diagnosis, prevent, and treat. For many years, the only available agents were amphotericin B and the azole antifungals. However, over the past decade several new antifungal agents with novel mechanisms of action, broader spectrums of activity, and fewer side effects have emerged. This article will briefly review the mechanisms of action, spectrum of activity, pharmacodynamics, pharmacokinetics, and safety of the most commonly used antifungal agents in clinical practice.
Azoles
For over two decades, the azole antifungals have been used in clinical practice to treat various fungal infections (TABLE 1). They are categorized into two distinctive classes: the imidazoles and the triazoles. The imidazoles include several agents, most notably clotrimazole, ketoconazole, and miconazole. The triazoles include fluconazole, itraconazole, terconazole, voriconazole, and posaconazole. As a class, they exert their effect by impairing the synthesis of ergosterol, a vital component in the fungal cellular membrane. This effect occurs through the inhibition of CYP450, which converts lanosterol to ergosterol, resulting in increased cellular permeability and leakage of cellular contents as well as inhibition of fungal growth.7,8 Generally, side effects include gastrointestinal (GI) disturbances (e.g., nausea, vomiting, diarrhea), hepatotoxicity, and rash. Since all of these agents are hepatically metabolized, many are potent CYP450 inhibitors.
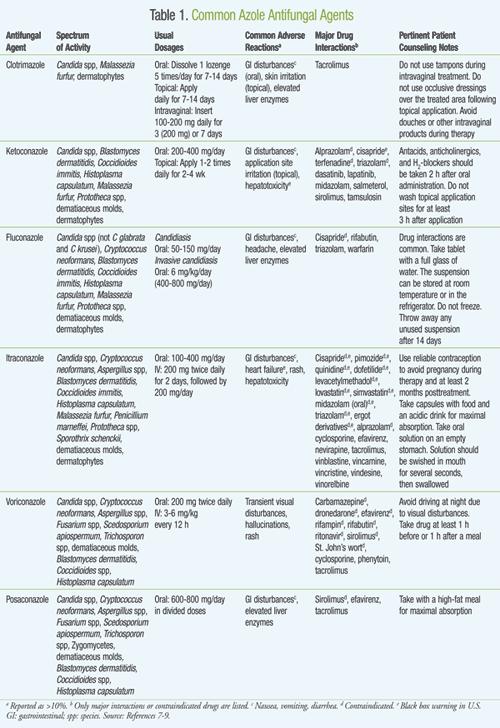
Imidazoles: The imidazoles are available in a variety of topical applications such as creams, lotions, shampoos, vaginal tablets, lozenges, and solutions that are mainly used to treat vaginal and epidermal candidiasis. Ketoconazole, the first orally available azole antifungal for the treatment of superficial and systemic fungal infections, has activity against a variety of Candida spp.9 In addition, it can be used to treat infections caused by dermatophytes, Malassezia furfur, and some dimorphic fungi, such as Blastomyces dermatitidis and Coccidioides spp.9 Absorption of ketoconazole from the GI tract is variable.8 For better absorption, patients who are prescribed ketoconazole tablets should be encouraged to take them with an acidic drink such as carbonated soda. Besides the general azole class adverse reactions, ketoconazole also carries a black box warning for hepatotoxicity and has significant drug–drug interactions, which further limits its use, and therefore should only be used as an alternative to safer, less toxic triazoles.7-9
Triazoles: Fluconazole, a first-generation triazole antifungal, has several advantages over other azole agents. These advantages include having a broader spectrum of activity, nearly 100% bioavailability because it does not undergo first-pass metabolism, and a variety of dosage forms that are not affected by the absence or presence of gastric acid.7,10 Fluconazole is active against the majority of Candida spp (except C krusei and C glabrata), as well as Cryptococcus neoformans, some dimorphic fungi, M furfur, Prototheca spp, dematiaceous molds, and dermatophytes.9,11 Fluconazole is an inhibitor of isoenzymes CYP2C9, CYP2C19, and CYP3A4; therefore, clinicians must perform an active medication review on patients prescribed fluconazole to avoid any potential drug interactions.
Similar to fluconazole, itraconazole can also be given orally or intravenously. Its spectrum of activity is similar to fluconazole’s, but it also has activity against Aspergillus spp, dematiaceous molds (e.g., Alternaria, Bipolaris, Curvularia), and Sporothrix schenckii.9 Due to its inotropic effects, itraconazole’s labeling includes a black box warning in patients with heart failure, particularly in patients receiving a total daily oral dose of 400 mg.7-9 Like fluconazole, it is a potent inhibitor of the CYP450 enzyme system. Since the various dosage formulations of itraconazole have different bioavailabilities, patients should be counseled on appropriate drug administration. For instance, itraconazole oral capsules should be taken with food and a high acidic beverage, as an acidic environment increases its absorption.7
Voriconazole, approved by the FDA in 2002, also has a broad spectrum of activity. It has activity against Aspergillus spp including amphotericin B–resistant Aspergillus terreus.9 Additional coverage includes more resistant strands of Candida spp, Fusarium spp, Scedosporium apiospermum, Trichosporon spp, and various molds.9 Voriconazole does not require an acidic environment for appropriate absorption to occur; thus, it has better bioavailability compared to ketoconazole or itraconazole. Voriconazole must be taken 1 hour before or 1 to 2 hours after a meal, as high-fat meals decrease its absorption.7-9 About 20% of patients experience transient visual disturbances such as photophobia, blurred vision, or color changes following either oral or IV administration.7,12 Dose-related hallucinations have also been reported in 15% of patients.7,12 While oral formulations are hepatically metabolized by CYP450 isoenzymes CYP2C19, CYP2C9, and CYP3A4, its affinity is greater for CYP2C19. The excretion of voriconazole is not affected by renal failure. However, the parenteral preparation is solubilized in a compound that is secreted by the kidneys, necessitating dose adaptation in cases of renal impairment, and it is contraindicated in those with an estimated creatinine clearance (CrCl) <50 mL/min. Voriconazole is associated with many drug–drug interactions. These interactions are mainly due to voriconazole’s inhibition of CYP2C19, CYP2C9, and CYP3A4. When administering voriconazole, patients should be instructed to take the drug on an empty stomach, as high-fat foods interfere with absorption.7-9
Posaconazole, the newest azole antifungal, was approved in 2006 and has a wide-ranging antifungal activity, including Candida spp resistant to older azoles.7,9 Unlike voriconazole, it also has activity against zygomycetes.13 Posaconazole is only available as an oral suspension that has poor bioavailability. However, if administered with a high-fat meal, posaconazole’s bioavailability is increased by 400%.14,15 The most commonly experienced adverse effects include GI disturbances and a potential for an elevation in liver enzymes.7-9
Polyenes
The polyene class of antifungals includes amphotericin B, nystatin, and natamycin. Before the introduction of broad-spectrum azoles and the echinocandins, amphotericin B was the standard of care for many systemic fungal infections, in spite of its toxicity risks. Polyenes exert their effects by disrupting the fungal cell membrane through binding to ergosterol, a component of the cell wall. This action results in increased cellular permeability and leakage of cellular contents, as well as inhibition of fungal growth.7 Amphotericin B is available in several formulations, including multiple liposomal parenteral formulations designed to increase tolerability. The lipid carriers of each formulation differ greatly, but these differences have no effect on therapeutic outcome and only confer a different protection against amphotericin adverse effects. AmBisome is a spherical carrier that contains amphotericin on the inside and outside of the vesicle, while Abelcet consists of amphotericin B complexed with two phospholipids in a 1:1 drug-to-lipid molar ratio.16,17 Amphotec contains amphotericin B complexed with cholesteryl sulfate, which upon reconstitution forms a colloidal dispersion of microscopic disk-shaped particles.18
Amphotericin B has activity against the majority of invasive fungi, including Candida spp, Aspergillus spp, and dimorphic fungi.9 Common toxicities include nephrotoxicity, hematologic toxicity, and infusion-related reactions.7-9 Because of the many adverse effects associated with amphotericin B, pharmacists can play a critical role in monitoring tolerability for patients. Premedication with acetaminophen and heparin are common measures taken to prevent infusion-related reactions such as fever, headache, and thrombophlebitis. Administering normal saline before the initiation of therapy can decrease drug-induced nephrotoxicity. In addition, avoiding other nephrotoxins, switching to other formulations of amphotericin, and correcting electrolyte abnormalities such as hypokalemia and hypomagnesemia are all means whereby pharmacists can assist in reducing adverse events.
Structurally, nystatin is closely related to amphotericin B, yet it is not given parenterally due to toxicity.7 It is usually fungistatic in vivo but may have fungicidal activity at high concentrations or against extremely susceptible organisms. It is available in oral and topical forms and has no significant drug interactions due to its lack of absorption from the gut. Adverse effects are infrequent, but in large doses it can produce mild and transient nausea, vomiting, diarrhea, and abdominal pain.7
Allylamines
Terbinafine is an allylamine antifungal that exerts its effects by inhibiting the enzyme squalene monooxygenase, a key enzyme in sterol biosynthesis in fungi. It is administered either topically or orally and is often used as a first-line agent for treating onychomycosis, a fungal nail infection. It is only active in vivo against dermatophytes and does not treat Candida or mold species.19 Terbinafine is available as a prescription and in OTC creams. It is not recommended for use in patients with chronic or active liver disease, and patients’ liver enzymes should be monitored before beginning treatment, even in those without pre-existing liver disease. Changes in patients’ ocular lenses and retinas have also been observed, but clinical significance of these changes is unknown. The manufacturer also recommends monitoring CBCs in patients planning on using terbinafine for >6 weeks due to decreases in absolute lymphocyte count observed in clinical trials.19
Echinocandins
The echinocandin class of antifungals is one of the newer classes, and it exerts its effects through inhibiting the synthesis of (1,3)-beta-d-glucan synthase, a vital component of the cell walls of various fungi, resulting in osmotic instability and ultimately fungal cell death.20 The novel mechanism of action of this class is very different from that of other antifungal classes, and it provides new therapeutic options for clinicians. The cell wall of C neoformans, however, consists mainly of alpha-(1,3)- or alpha-(1,6)-glucan, thus rendering it resistant to the echinocandin class. This antifungal class includes caspofungin, micafungin, and anidulafungin (TABLE 2). All have similar spectrums of activity and are only available intravenously. Furthermore, each of the echinocandins has an excellent safety profile, as most of the adverse effects involve infusion-related reactions.20
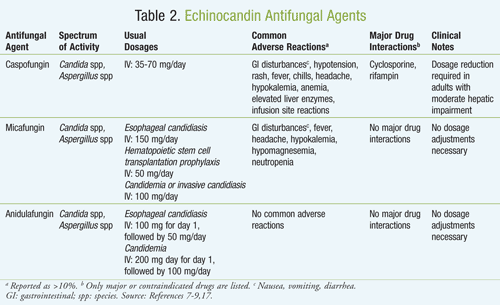
Caspofungin was approved in 2001 for the treatment of patients with invasive aspergillosis who cannot tolerate or who are refractory to other antifungal treatments. It is also approved for treatment of esophageal candidiasis, intra-abdominal abscesses, peritonitis, and pleural space infections caused by Candida spp.9 Empirically, it is used for the treatment of fever of unknown origin in neutropenic patients. Caspofungin does not substantially interfere with the CYP450 enzyme system, but it does undergo significant hepatic metabolism. Caution should be used in patients with hepatic disease, as dosage adjustments may become necessary.21
Micafungin became available in 2005 and was approved for the treatment of esophageal candidiasis as well as for prophylaxis in patients undergoing stem cell transplantation. Micafungin is highly protein bound (>99%), primarily to albumin.7 At therapeutically relevant concentrations, micafungin does not competitively displace bilirubin binding to albumin; therefore, it does not cause kernicterus (brain damage caused by excessive jaundice). Micafungin also has relatively few drug–drug interactions since it is a weak inhibitor of CYP3A4.
In 2006, the FDA approved anidulafungin for the treatment of esophageal candidiasis, candidemia, peritonitis, and intra-abdominal abscesses due to Candida spp. Anidulafungin is not hepatically metabolized and is not a clinically relevant substrate, inducer, or inhibitor of CYP450 enzymes. Uniquely, anidulafungin undergoes slow chemical degradation that takes place at a physiologic temperature and pH rather than metabolism.22 Degradation products pass into the feces via the biliary tree and very small amounts are also found in the urine.22 Therefore, patients who may have renal or hepatic insufficiency do not require dosage adjustments.7-9
Summary
With the evolving changes in antifungal therapy, treatment of fungal infections has become more manageable. Although resistance is on the rise, special precautions to reduce the overuse of antifungals, broad-spectrum antibiotics, and other predisposing factors should be followed in order to slow the progression of resistance and enhance patient outcomes. Recognizing those patients at increased risk for developing fungal infections, especially invasive fungal infections, will aid in improving morbidity and mortality associated with these infectious diseases. With a number of antifungal agents available, it is important for pharmacists to be aware of each drug’s characteristics so that they are better equipped to make sound therapeutic decisions in the treatment of various fungal infections.
The authors would like to thank Melissa Diamond and Dee Dugan for their contributions.
REFERENCES
1. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
2. Lai CC, Tan CK, Huang YT, et al. Current challenges in the management of invasive fungal infections. J Infect Chemother. 2008;14:77-85.
3. Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546-1554.
4. Pappas PG, Rex JH, Lee J, et al. A prospective observational study of candidemia: epidemiology, therapy, and influences on mortality in hospitalized adult and pediatric patients. Clin Infect Dis. 2003;37:634-643.
5. Tortorano AM, Peman J, Bernhardt H, et al. Epidemiology of candidaemia in Europe: results of 28-month European Confederation of Medical Mycology (ECMM) hospital-based surveillance study. Eur J Clin Microbiol Infect Dis. 2004;23:317-322.
6. Pappas PG, Kauffman CA, Andes D, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:503-535.
7. Clinical Pharmacology Web site [subscription required]. http://cpip.gsm.com.ezproxy.
8. Micromedex Healthcare Series Web site [subscription required]. www.thomsonhc.com.ezproxy.
9. Lexi-Comp Web site [subscription required]. www.crlonline.com.ezproxy.
10. Diflucan (fluconazole) package insert. New York, NY: Pfizer, Inc; March 2008.
11. Ostrosky-Zeichner L, Rex JH, Pappas PG, et al. Antifungal susceptibility survey of 2,000 bloodstream Candida isolates in the United States. Antimicrob Agents Chemother. 2003;47:3149-3154.
12. Hoffman HL, Rathbun RC. Review of the safety and efficacy of voriconazole. Expert Opin Investig Drugs. 2002;11:409-429.
13. Sun QN, Fothergill AW, McCarthy DI, et al. In vitro activities of posaconazole, itraconazole, voriconazole, amphotericin B, and fluconazole against 37 clinical isolates of zygomycetes. Antimicrob Agents Chemother. 2002;46:1581-1582.
14. Courtney R, Wexler D, Radwanski E, et al. Effect of food on the relative bioavailability of two oral formulations of posaconazole in healthy adults. Br J Clin Pharmacol. 2004;57:218-222.
15. Courtney R, Radwanski E, Lim J, et al. Pharmacokinetics of posaconazole coadministered with antacid in fasting or nonfasting healthy men. Antimicrob Agents Chemother. 2004;48:804-808.
16. AmBisome (amphotericin B) package insert. Deerfield, IL: Gilead Sciences, Inc; October 2008.
17. Abelcet (liposome) package insert. Bridgewater, NJ: Enzon Pharmaceuticals, Inc; March 1998.
18. Amphotec (amphotericin B cholesteryl sulfate complex) package insert. Bedford, OH: Ben Venue Laboratories, Inc; July 2005.
19. Lamasil (terbinafine) package insert. East Hanover, NJ: Novartis Pharmaceuticals; November 2005.
20. Onishi J, Meinz M, Thompson J, et al. Discovery of novel antifungal (1,3)-beta-d-glucan synthase inhibitors. Antimicrob Agents Chemother. 2000;44:368-377.
21. Cancidas (caspofungin) package insert. Whitehouse Station, NJ: Merck & Co, Inc; 2010.
22. Dowell JA, Knebel W, Ludden T, et al. Population pharmacokinetic analysis of anidulafungin, an echinocandin antifungal. J Clin Pharmacol. 2004;44:590-598.
To comment on this article, contact rdavidson@uspharmacist.com.


