US Pharm. 2013;38(8):HS11-HS16.
ABSTRACT: Uremic syndrome involves the impairment of several biochemical and physiological functions associated with deteriorating renal function. Uremia presents as a broad array of symptoms and is characterized by an accumulation of toxins, which are classified based on size and protein binding. Dialysis is the mainstay of toxin removal, although kidney transplantation may be necessary in severe cases. Clinical effects of uremic toxins are proposed based on surrogate mechanisms of oxidative, endothelial, and erythrocyte damage. Currently, drug therapy targeting uremic symptoms is anecdotal. Uremic bleeding is the most well-described symptom in the literature, and viable therapeutic options for management are available.
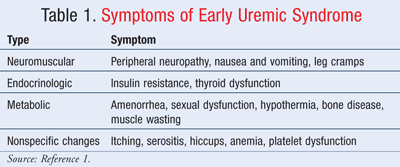
Uremic patients present with various signs and symptoms collectively referred to as uremic syndrome. This syndrome involves the impairment of several biochemical and physiological func-tions associated with deteriorating renal function. Symptoms are nonspecific and difficult to identify in early disease (TABLE 1).1 Cardinal signs and symptoms of advanced disease include anorexia and eventual weight loss, confusion, lethargy, bleeding, coma, and death.2
Treatment of uremia primarily involves dialysis (hemodialysis and peritoneal) and ultimately kidney transplantation for eligible candi-dates. Inadequate removal of uremic toxins through conventional dialysis may result in a phenomenon known as residual syndrome. Patients with this syndrome may experience subtle signs of malnutrition, mild lethargy, infection, and serositis.2 Complete symptom reversal usually requires kidney transplantation. Although transplantation is the most effective method of treatment, it is increasingly difficult due to the limited number of donor kidneys.3
Uremic Solutes
Uremia is characterized by an accumulation of toxins in chronic kidney disease (CKD) and end-stage renal disease that leads to illness. Generally, uremic symptoms cannot be attributed to changes in volume status, electrolyte disturbances, or absence of renal by-products.1
Although predominant, urea is only one of many toxins that accumulate in CKD. Other toxic solutes include peptides and small proteins, guanidines, phenols, indoles, aliphatic amines, furans, polyols, nucleosides, dicarboxylic acids, and carbonyls.1,4 Classification of uremic toxins is based on several criteria, one being that removal of the toxin demonstrates a reduction in symptoms. Inability to effectively remove certain solutes to demonstrate resolution of symptoms can make toxin assignment difficult. Data linking toxic effects and disturbances of various biological and biochemical functions associated with specific uremic solutes are limited.
The European Uremic Toxin Work Group has identified over 100 uremic toxins and has classified them by structure.4,5 Uremic compounds are categorized into three groups: 1) small, water-soluble, nonprotein-bound compounds (<500 D); 2) small, lipid-soluble and/or protein-bound compounds; and 3) large “middle” molecules.5 Accumulated toxic solutes in uremia produced by gut bacteria and mammalian cells are positively charged compounds with preferential intracellular distribution, making removal by dialysis difficult.1,2 Protein-bound molecules and large middle molecules are also inadequately removed by current dialysis methods. Continued strides to improve dialysis strategies and develop novel approaches to inhibit toxin production may lead to greater symptomatic relief and survival benefits in CKD.
Small, Water-Soluble Compounds: In kidney failure, urea was the first organic compound to be identified in high concentrations systemically.1 Urea is a water-soluble 60-D solute that is minimally toxic and only partially accounts for some uremic symptoms.6 Studies show mixed results and poor correlation of symptom resolution and improved survival with urea removal through dialysis.1,6 Additionally, urea, which used to be the prototype of small, water-soluble compound retention and dialysis removal, does not appear to adequately represent the behavior exhibited by similar compounds during hemodialysis.6
Guanidines are water-soluble uremic compounds structurally similar to urea with pleiotropic effects, including neurotoxicity.5,6 Recently, guanidines have been investigated for having potential cardiovascular effects in uremic patients. High concentrations of guanidines are believed to activate leukocyte function, leading to proinflammatory production of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6).6 The impact of guanidines on albumin may also increase unbound levels of homocysteine, a protein-bound compound linked to cardiovascular damage through macrophage activation, resulting in high superoxide anion levels.7 Folic acid can counteract homocysteine elevation to some degree.7 Guanidines exhibit large volumes of distribution, hindering their removal via hemodialysis, but longer durations or increasing frequency of hemodialysis sessions can potentially improve their removal.6
Protein-Bound Molecules: P-cresol is a 108-D lipophilic phenol prototype of protein-bound molecules produced by intestinal bacteria.4,5 High concentrations of p-cresol have been associated with increased hospitalization due to infections, uremic symptoms, mortality, and cardiovascular disease.6 However, p-cresol is now known to be absent in the body, and its appearance in uremic samples is a result of artifact from hydrolysis of conjugates, such as p-cresyl sulfate (PCS), a compound present in vivo.5 P-cresol attenuates leukocyte activity, whereas PCS improves leukocyte activity.5 Recent studies with PCS produced findings of increased cardiovascular disease and mortality similar to those of p-cresol despite having opposing effects.6 Removal of p-cresol is no better with high-flux than with low-flux dialysis.5 Fractionated plasma separation adsorption could enhance p-cresol removal; however, studies show evidence of severe coagulation manifestations.6
Indols are protein-bound molecules also linked to various mechanisms of endothelial damage.5 They may interfere with protein binding of acidic drugs and inhibit tubular secretion of highly protein-bound drugs, increasing the risk of drug toxicity.6 Appearance of indols requires intestinal bacteria metabolism; thus, administration of oral prebiotics such as bifidobacteria in gastro-resistant capsules may reduce indoxyl sulfate levels.6 Dialysis is usually a poor strategy for removal of protein-bound indols.
Large “Middle” Molecules: Middle molecules are arbitrarily assigned as compounds >500 D in molecular weight and include toxins such as TNF-α and IL-6. Middle molecules are believed to be involved in inflammation, endothelial destruction, smooth-muscle cell proliferation, and coagulation, although studies determining the nature of their effect in uremic processes are lacking.6
Beta2-microglobulin is 12,000 D and the prototype for middle-molecule uremic toxins. It is used as a marker in peripheral vascular disease and may play a role in arterial stiffness and bone disease.6 Beta2-microglobulin is more readily cleared by high-flux hemodialysis, which incorporates greater permeability and larger pore size for more effective solute removal.1 Removal of beta2-microglobulin in CKD has been associated with good outcomes and improved mortality related to infection.6
Leptin is a large, protein-bound, 16-KD molecule that may provoke vascular damage.4,7 It increases tissue factor expression linked to clotting and inflammation and may enhance atherosclerosis in CKD.7
Pharmacologic Therapy of Uremic Toxins
Current cardiovascular therapies proportionally prevent more cardiovascular death in patients without CKD versus those with renal failure. Determining adverse biological effects caused by toxins that accumulate in uremia can provide strategies to improve quality of life and reduce mortality in patients with CKD. Currently, what is known is that many of these designated toxins impair functions in leukocytes, endothelium, and smooth muscle cells, which can subsequently contribute to immune deficiency, inflammation, atherosclerosis, and cardiovascular disease.7
The esoteric effects of uremia on vasculature and immunity make it difficult to isolate mechanisms by which drugs may improve uremic symptoms. However, drugs that have been used empirically to counteract toxic effects of uremia include aspirin for antiplatelet and anti-inflammatory properties; antihypertensives such as angiotensin-converting enzyme (ACE) inhibitors, beta-adrenergic blockers, and diuretics to normalize blood pressure; statins to lower atherosclerotic cholesterol; phosphate binders to lower phosphorous levels; and folic acid to reduce homocysteine levels.7 Future targets indicate a role for drugs that inhibit culprit receptors, calcium transporters, transcription factors, and promoters of oxidative stress that are specific to the detrimental effects of uremia.7
Uremic Bleeding
Among the numerous symptoms of uremia in CKD, uremic bleeding is perhaps the most well-documented complication.8-10 Although its exact pathophysiology is unknown, platelet dysfunction seems to play the largest role and involves impairment of both platelet aggregation and adhesive-ness.8,11,12 Uremic toxins, elevated prostaglandin I2 (PGI2) levels, increased nitric oxide (NO) production, von Willebrand factor (vWF) abnormalities, and anemia are among the leading causes of hemostatic disturbance in uremic patients.8-12
With regard to urea, there does not seem to be a clear relationship between blood urea nitrogen (BUN) levels and abnormal bleeding time in renal failure patients.13 However, excess levels of urea can ultimately result in greater formation of guanidinosuccinic acid (GSA), which may inhibit adenosine diphosphate (ADP)–induced platelet aggregation. Phenolic acids also inhibit this aggregation, contributing to the platelet dysfunction.14,15
Elevated levels of GSA and methylguanidine have been implicated in the stimulation of NO production, resulting in platelet adhesion and aggregation dysfunction.9,16,17 Elevated NO levels stimulate guanylyl cyclase and result in excess cyclic guanosine monophosphate (cGMP). This leads to a reduction in thromboxane A2 and ADP levels, further contributing to abnormal platelet aggregation.10,14,15-17 An elevation of PGI2 may also be seen in chronic renal failure patients (with abnormal bleeding times) due to reduced levels of thromboxane A2 and ADP, which result from adenylyl cyclase stimulating the production of cyclic adenosine monophosphate (cAMP).8,18,19 PGI2 may also play a role in the inhibition of platelet spreading, thereby reducing adhesion and thrombus formation.20
An abnormal interaction between vWF and glycoprotein (GP) Ib/IX may also lead to platelet dysfunction in uremic patients.10,21 Without appropriate binding of vWF to these receptors, levels of thromboxane A2 and ADP are ultimately reduced and GPIIb/IIIa receptors are not activated, leading to additional platelet aggregation issues.22,23
In patients with anemia associated with chronic renal failure, decreased levels of erythropoietin and red blood cells may also result in abnormal platelet aggregation.11 At normal levels, red blood cells release thromboxane A2 and ADP and allow platelets to adhere to endothelial surfaces and form platelet plugs in response to injury.11,24,25 Low levels of hemoglobin can remove and inactivate NO and may also contribute to platelet dysfunction.26
Management of Uremic Bleeding
Patients with uremic bleeding may present with various symptoms of bleeding (e.g., epistaxis, ecchymosis), as well as mild thrombocytopenia.10 Other causes of bleeding must be ruled out prior to formulating a therapeutic plan for uremic patients. Bleeding time is considered to be the preferred test for assessing clinical bleeding.10,13
Dialysis: In patients with advanced renal impairment, dialysis may be necessary to remove by-products and uremic toxins. Approximately two-thirds of uremic patients with bleeding may exhibit partial bleeding-time correction with hemodialysis or peritoneal dialysis.27-29 In one study, bleeding time was normalized during 30% of dialysis sessions.28 Hemodialysis without anticoagulation is recommended in patients with active bleeding.30 In addition, certain small, water-soluble guanidino compounds may require longer or more frequent dialysis sessions for their removal (due to their higher distribution volumes as compared to urea).31
Desmopressin: This vasopressin analogue stimulates the release of vWF from endothelial cells, as well as the release of factor VIII (a protein essential for blood clotting) from other storage sites.32-34 It may also aid in the expression of glycoprotein on platelet membranes.34 Improvement in bleeding time can be seen within 1 hour of administration, and duration is approximately 4 to 8 hours, with bleeding time generally returning to baseline within 24 hours.10,35
Although not approved for uremic bleeding, studies have demonstrated a normalization or reduction in bleeding time with desmopressin. Doses of desmopressin in these studies ranged from 0.3 mcg/kg to 0.4 mcg/kg IV or SC.35-38 Adverse events included headache, flushing, and rare thrombotic events. Hyponatremia and reduced urine volume may also occur. In addition, tachyphylaxis after one or two doses may develop and is believed to be a result of vWF or factor VIII storage depletion.8,35-38
Cryoprecipitate: Cryoprecipitate is a blood product containing factor VIII, vWF, and fibrinogen, which may contribute to platelet aggregation in patients with uremic bleeding.10,39,40 Ten units/bags of American Red Cross–prepared cryoprecipitate administered IV over 30 minutes have resulted in decreased bleeding times within 4 to 12 hours of infusion, with an onset of approximately 1 hour.10,39,41 However, this product is generally used in patients who do not respond to desmopressin or have a contraindication to desmopressin therapy due to the potential risks of infection transmission, allergic or anaphylactic reactions, and potential serologic incompatibilities associated with cryoprecipitate.10,40,41
Erythropoietin: Elevations in hemoglobin ≥10 g/dL may improve platelet function and reduce bleeding times in patients with anemia of CKD.42,43 Recombinant erythropoietin-stimulating agents (ESAs) have been seen to decrease or normalize bleeding time in uremic patients with a target hematocrit of >30%, which can take up to 9 weeks in uremic patients.10,42,44,45 However, an increase in reticulated platelets can be observed within 7 days of ESA initiation.42,45 In addition, ESAs may aid with platelet adhesion and aggregation in the acute setting.46 Transfusion of packed red blood cells is also commonly used acutely to correct anemia and for patients with active bleeding complications.40,43
Conjugated Estrogens: Conjugated estrogens are believed to decrease the production of NO47 and have yielded beneficial effects (i.e., bleeding-time and clinical bleeding improvements) in the chronic management of bleeding in uremic patients.10,40,48-51 Although oral and transdermal therapies have been shown to aid in the control of bleeding, the majority of evidence supports 0.6 mg/kg of IV-administered conjugated estrogens once daily for 5 days. The onset of action is approximately 6 hours, with a total duration of about 14 to 21 days.48-50 However, long-term use of conjugated estrogen therapy is limited due to estrogen-associated adverse effects.10,40
Conclusion
Uremia presents as a broad scope of symptoms appearing in CKD caused by an accumulation of toxins. Removal of larger and highly protein-bound molecules is difficult, even with improving methods of dialysis. From available reviews, detrimental clinical effects of uremic compounds can be assumed based on surrogate mechanisms of damage to leukocytes, endothelium, and erythrocytes exhibited by these solutes. Currently, there are no useful drugs to treat uremic symptoms aimed at specific toxins. The armamentarium of drugs used in CKD is similar to drugs used to prevent cardiovascular disease in the general population. Among the symptoms seen in uremia, uremic bleeding is the most well described in the literature, and viable therapeutic options for management have been elucidated.
REFERENCES
1. Meyer TW, Hostetter TH. Uremia. N Engl J Med. 2007;357:1316-1325.
2. Depner TA. Uremic toxicity: urea and beyond. Semin Dial. 2001;14:246-251.
3. USRDS 2012 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda, MD: U.S. Renal Data System; 2012.
4. Vanholder R, De Smet R, Glorieux G, et al; European
Uremic Toxin Work Group. Review on uremic toxins: classification,
concentration, and interindividual variability. Kidney Int. 2003;63:1934-1943.
5. Vanholder R, Van Laecke S, Glorieux G. What is new in uremic toxicity? Pediatr Nephrol. 2008;23:1211-1221.
6. Neirynck N, Vanholder R, Schepers E, et al. An update on uremic toxins. Int Urol Nephrol. 2013;45:139-150.
7. Vanholder R, Baurmeister U, Brunet P, et al; European Uremic Toxin Work Group. A bench to bedside view of uremic toxins. J Am Soc Nephrol. 2008;19:863-870.
8. Weigert AL, Schafer AI. Uremic bleeding: pathogenesis and therapy. Am J Med Sci. 1998;316:94-104.
9. Noris M, Remuzzi G. Uremic bleeding: closing the circle after 30 years of controversies? Blood. 1999;94:2569-2574.
10. Hedges, SJ, Dehoney SB, Hooper JS, et al. Evidence-based treatment recommendations for uremic bleeding. Nat Clin Pract Nephrol. 2007;3:138-153.
11. Malyszko J, Malyszko JS, Mysliwiec M, Buczko W. Hemostasis in chronic renal failure. Rocz Akad Med Bialymst. 2005;50:126-131.
12. Zwaginga JJ, IJsseldijk MJ, de Groot PG, et al.
Defects in platelet adhesion and aggregate formation in uremic bleeding
disorder can be attributed to factors in plasma. Arterioscler Thromb. 1991;11:733-744.
13. Steiner RW, Coggins C, Carvalho AC. Bleeding time in uremia: a useful test to assess clinical bleeding. Am J Hematol. 1979;7:107-117.
14. Horowitz HI, Stein IM, Cohen BD, White JG. Further
studies on the platelet-inhibitory effect of guanidinosuccinic acid and
its role in uremic bleeding. Am J Med. 1970;49:336-345.
15. Rabiner SF, Molinas F. The role of phenol and phenolic
acids on the thrombocytopathy and defective platelet aggregation of
patients with renal failure. Am J Med. 1970;49:46-51.
16. Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057-1058.
17. Marietta M, Fachinetti F, Neri I, et al. L-arginine
infusion decreases platelet aggregation through an intraplatelet nitric
oxide release. Thromb Res. 1997;228:229-235.
18. Remuzzi G, Cavenaghi AE, Mecca G, et al. Prastacyclin-like activity and bleeding in renal failure. Lancet. 1977;310:1195-1197.
19. Boccardo P, Remuzzi G, Galbusera M. Platelet dysfunction in renal failure. Sem Thromb Hemost. 2004;30:579-589.
20. Weiss HJ, Turitto VT. Prostacyclin (prostaglandin I2, PGI2) inhibits platelet adhesion and thrombus formation on subendothelium. Blood. 1979;53:244-250.
21. Mohri H, Fujimura Y, Shima M, et al. Structure of the von Willebrand factor domain interacting with glycoprotein Ib. J Biol Chem. 1988;263:17901-17904.
22. Savage B, Shattil SJ, Ruggeri ZM. Modulation of
platelet function through adhesion receptors: a dual role for
glycoprotein IIb-IIIa (integrin alpha IIb beta 3) mediated by fibrinogen
and glycoprotein Ib-von Willebrand factor. J Biol Chem. 1992;267:11300-11306.
23. Harker LA, Hanson SR, Kelly AB. Antithrombotic
strategies targeting thrombin activities, thrombin receptors and
thrombin generation. Thromb Haemost. 1997;78:736-741.
24. Jacobson LO, Goldwasser E, Fried W, Plzak L. Role of the kidney in erythropoiesis. Nature. 1957;179:633-634.
25. Valles J, Santos MT, Aznar J, et al. Erythrocytes
metabolically enhance collagen-induced platelet responsiveness via
increased thromboxane production, adenosine diphosphate release, and
recruitment. Blood. 1991;78:154-162.
26. Martin W, Villani GM, Jothianandan D, Furchgott RF.
Blockade of endothelium-dependent and glyceryl trinitrate-induced
relaxation of rabbit aorta by certain ferrous hemoproteins. J Pharmacol Exp Ther. 1985;233:679-685.
27. Lindsay RM, Friesen M, Aronstam A, et al. Improvement of platelet function by increased frequency of hemodialysis. Clin Nephrol. 1978;10:67-70.
28. Stewart JH, Castaldi PA. Uraemic bleeding: a reversible platelet defect corrected by dialysis. Q J Med. 1967;36:409-423.
29. Nenci GG, Berrettini M, Agnelli G, et al. Effect of
peritoneal dialysis, haemodialysis and kidney transplantation on blood
platelet function. I. Platelet aggregation by ADP and epinephrine. Nephron. 1979;23:287-292.
30. Galbusera M, Remuzzi G, Boccardo P. Treatment of bleeding in dialysis patients. Semin Dial. 2009;22:279-286.
31. Eloot S, Torremans A, De Smet R, et al. Kinetic
behavior of urea is different from that of other water-soluble
compounds: the case of the guanidino compounds. Kidney Int. 2005;67:1566-1575.
32. Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Blood. 1997;90:2515-2521.
33. Zeigler ZR, Megaludis A, Fraley DS. Desmopressin
(d-DAVP) effects on platelet rheology and von Willebrand factor
activities in uremia. Am J Hematol. 1992;39:90-95.
34. Gordz S, Mrowietz C, Pindur G, et al. Effect of
desmopressin (DDAP) on platelet membrane glycoprotein expression in
patients with von Willebrand’s disease. Clin Hemorheol Microcirc. 2005;32:83-87.
35. Köhler M, Hellstern P, Tarrach H, et al. Subcutaneous
injection of desmopressin (DDAVP): evaluation of a new, more
concentrated preparation. Haemostasis. 1989;19:38-44.
36. Vigano GL, Mannucci PM, Lattuada A, et al. Subcutaneous desmopressin (DDAVP) shortens the bleeding time in uremia. Am J Hematol. 1989;31:32-35.
37. Mannucci PM, Remuzzi G, Pusineri F, et al. Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. N Engl J Med. 1983;308:8-12.
38. Watson AJ, Keogh JA. Effect of 1-deamino-8-D-arginine vasopressin on the prolonged bleeding time in chronic renal failure. Nephron. 1982;32:49-52.
39. Janson PA, Jubelirer SJ, Weinstein MJ, Deykin D. Treatment of the bleeding tendency in uremia with cryoprecipitate. N Engl J Med. 1980;303:1318-1320.
40. Salman S. Uremic bleeding: pathophysiology, diagnosis, and management. Hosp Physician. 2001;76:45-50.
41. Triulzi DJ, Blumberg N. Variability in response to cryoprecipitate treatment for hemostatic defects in uremia. Yale J Biol Med. 1990;63:1-7.
42. Cases A, Escolar G, Reverter JC, et al. Recombinant
human erythro-poietin treatment improves platelet function in uremic
patients. Kidney Int. 1992:42;668-672.
43. Livio M, Gotti E, Marchesi D, et al. Uraemic bleeding: role of anaemia and beneficial effects of red cell transfusions. Lancet. 1982;2:1013-1015.
44. Vigano G, Benigni A, Mendogni D, et al. Recombinant human erythropoietin to correct uremic bleeding. Am J Kidney Dis. 1991;18:44-49.
45. Zwaginga JJ, IJsseldijk MJ, de Groot PG, et al.
Treatment of uremic anemia with recombinant erythropoietin also reduces
the defects in platelet adhesion and aggregation caused by uremic
plasma. Thromb Haemost. 1991;66:638-647.
46. Tassies D, Reverter JC, Cases A, et al. Effect of
recombinant human erythropoietin treatment on circulating reticulated
platelets in uremic patients: association with early improvement in
platelet function. Am J Hematol. 1998;59:105-109.
47. Zoja C, Noris M, Corna D, et al. L-arginine, the
precursor of nitric oxide, abolishes the effect of estrogen on bleeding
time in experimental uremia. Lab Invest. 1991;65:479-483.
48. Livio M, Mannucci PM, Vigano G, et al. Conjugated estrogens for the management of bleeding associated with renal failure. N Engl J Med. 1986;315:731-735.
49. Vigano G, Gaspari F, Locatelli M, et al. Dose-effect
and pharmaco-kinetics of estrogens given to correct bleeding time in
uremia. Kidney Int. 1988;34:853-858.
50. Heistinger M, Stockenhuber F, Schneider B, et al.
Effect of conjugated estrogens on platelet function and prostacyclin
generation in CRF. Kidney Int. 1990;38:1181-1186.
51. Sloand JA, Schiff MJ. Beneficial effect of low-dose transdermal estrogen on bleeding time and clinical bleeding in uremia. Am J Kidney Dis. 1995;26:22-26.
To comment on this article, contact rdavidson@uspharmacist.com.



