US Pharm. 2012;37(8):31-34.
West Nile virus (WNV), a member of the Flaviviridae family, is most commonly found in West Asia, Africa, Europe, and the Middle East. This RNA virus can infect the central nervous system (CNS) of various hosts, causing severe neurologic disease.1 It was initially discovered in 1937 in the West Nile district of Uganda.2 Historically, WNV has been associated with infrequent human outbreaks associated with asymptomatic or mild febrile illness that was self-limiting, affecting mostly children, groups of soldiers, and healthy adults in Israel and Africa. Since the mid-1990s, however, there has been an increase in human and equine outbreaks and an increase in severe human disease.1-4
Prior to the 1990s, WNV had never been detected in North America. The first documented occurrence of WNV in the Western Hemisphere was during an outbreak of encephalitis in Queens, New York, in the late summer of 1999.5 Since this time, WNV has spread from one state in 1999, to three states in 2000, 10 states in 2001, and 46 states in 2003.6 It has been detected in all 48 states in the continental United States, the District of Columbia, and Puerto Rico.7 WNV has also spread northward and southward, affecting Canada, Latin America, and the Caribbean.8,9 WNV has become an emerging infectious disease in the Western Hemisphere, and is the most common cause of epidemic meningoencephalitis in this region.2 This article will review the epidemi-ology, clinical presentation, diagnosis, current treatment, and prevention of WNV.
Epidemiology
WNV is transmitted to humans through the bite of infected Culex mosquitoes; mosquitoes become infected after feeding on vertebrate amplifying hosts, most commonly birds.10,11 When the virus was first identified in North America, several common mosquito species were implicated in the transmission cycle.11 Since then, the virus has been identified in 64 mosquito species in the U.S.12 Survival of the virus in the U.S. has been mediated by the Culex mosquitoes; WNV is transmitted from female Culex mosquitoes to the next generation, and as adults these mosquitoes hibernate in the winter, allowing for persistence of the virus through the winter and reemergence in the spring.9,11
Peak transmission of WNV in North America occurs between July and October, but cases have been reported as early as April and as late as December.2,9 In more tropical climates, year-round transmission is possible.4 Human transmission is dependent on the abundance and feeding patterns of infected mosquitoes, as well as human exposure to the mosquitoes.10 During epidemics in Africa, up to 55% of the affected population has become infected; however, less than 3% of the affected U.S. population has been infected during times of epidemics.10,13 It is thought that this level of infection is too low to affect the frequency of epidemics or to alter their intensity through protective immunity.10
Human-to-human transmission of WNV does not generally occur; humans, horses, and most other mammals do not develop high-level viremia. Because of this, humans, horses, and most other mammals were considered to be dead-end hosts, being unable to transmit the virus through mosquito bites.6 This thought changed when 23 cases of WNV infections were reported after blood transfusions in 2002.14 This led to the FDA approval and implementation of WNV nucleic acid amplification tests to screen donor blood and prevent the spread of WNV via blood products. Other routes of WNV transmission that were first reported in 2002 included organ transplantation, intrauterine means, and possibly breastfeeding.10 In addition, WNV has been occupationally acquired by laboratory workers through percutaneous inoculation and possibly through aerosol exposure.10
Risk Factors
People of all ages are susceptible to WNV infection, but the elderly are more vulnerable to neuroinvasive WNV disease and death; the risk is also slightly higher for males and immunosuppressed organ transplant recipients.15,16 It is not known whether other immunocompromised individuals are at an increased risk for development of severe WNV.9
A case-control study found no association between WNV meningoencephalitis and hypertension, other underlying medical conditions, or cigarette smoking.17 In New York City, it was found that age over 75 years and the presence of diabetes were independent risk factors for the seven deaths that occurred in 1999.5 It has been postulated that hypertension and cerebrovascular disease are important risk factors for severe disease by promoting virus entry and replication in the blood-brain barrier endothelium.11
Presentation
The majority of cases of WNV infection are asymptomatic, but infected persons can experience West Nile fever (WNF), meningitis, encephalitis, and acute flaccid paralysis.6,13 About 20% of patients will develop WNF, and less than 1% of those infected will develop neuroinvasive disease.13 The typical incubation period for WNV is 2 to 14 days.7,6,18,19
WNF is used to describe symptomatic infections without neuroinvasive disease.20 It is usually self-limiting, with most symptoms lasting 3 to 6 days; fatigue and muscle weakness can persist for as long as 28 days.18,20 The usual presentation of WNF is fever of sudden onset, headache, back pain, malaise, eye pain, anorexia, and maculopapular rash.21,22
Patients over the age of 50 years are at a greater risk for developing neuroinvasive disease, which clinically presents as meningitis, encephalitis, or acute flaccid paralysis.18 Fever is experienced by 70% to 100% of patients with WNV neuroinvasive disease, followed by headache, gastrointestinal symptoms, and rash.9, 23 Other symptoms include muscle aches, weakness, altered mental status, fatigue, back pain, and stiff neck.9
West Nile encephalitis (WNE) ranges in severity from a mild, confusional state to severe encephalopathy, coma, and death.24 Some patients experience a prodrome of fever, headache, and other nonspecific symptoms lasting from 1 to a few days; others experience an abrupt onset of fever along with signs of encephalitis.19 Movement disorders may occur, especially extrapyramidal disorders.24 Patients will develop a coarse tremor in the upper extremities, some may experience myoclonus, and features of parkinsonism may be seen.24 Patients have also been known to present with muscle weakness; general progression of muscle weakness in severity coupled with a change in the level of consciousness increases the risk factors for death due to WNV.4
Acute flaccid paralysis (AFP) can occur in 5% to 15% of patients with neuroinvasive disease and can cause symptoms ranging from single-extremity weakness to quadriplegia, including paralysis of the respiratory muscles.22,23 Presentation of symptoms can be Guillain-Barré syndrome–like or poliomyelitis-like.22 The poliomyelitis-like AFP is more common, compromising 84% of WNV AFP cases.23 Younger patients may be more likely to experience AFP, but more deaths due to AFP occur primarily in the elderly.25,26
Some rare neurologic manifestations of WNV can include cranial nerve abnormalities, myelitis, optic neuritis, and seizures.21 Ocular manifestations include multifocal choroiditis, vitritis, and chorioretinitis; rare extraneurologic manifestations include myocarditis, pancreatitis, and hepatitis, as the affected organs are sites of high viral replication.19,21
Diagnosis
WNV should be part of the differential diagnosis for anyone who presents with an acute fever or neurologic illness and has recently been exposed to mosquitoes, lives in an enzootic area, or has received a recent blood transfusion or transplant.9 The most efficient way to diagnosis WNV is by detecting immunoglobulin M (IgM) antibody to the virus in the serum or cerebrospinal fluid (CSF) by using the IgM antibody capture enzyme-linked immunosorbent assay (MAC-ELISA). 4 Presence of IgM antibodies in CSF is indicative of CNS infection because this antibody does not cross the blood-brain barrier; at least 90% of patients who present with meningoencephalitis have detectable IgM antibody in the CSF within 8 days of symptom presentation.21
When interpreting the serologic results, it must be noted that false-positive results can occur in patients who have recently received the yellow fever or Japanese encephalitis vaccine or in those who have recently been infected with St. Louis encephalitis, yellow fever, dengue, or the Powassan virus; this is due to the close antigenic relationship with related flaviviruses.4,6,9 Any positive readings in these patients should be tested for specific neutralizing antibody.6 The plaque-reduction neutralization test (PRNT) is the most specific test for the arthropod-borne flaviviruses.4 Because WNV is asymptomatic in the majority of patients and IgM antibody can persist for 6 months or longer, diagnosing patients who live in endemic areas may be challenging. If these patients present with clinical illness, their MAC-ELISA most probably detected IgM antibody from a previous infection; an increase in the PRNT will confirm acute illness.
Prognosis
The overall case-fatality rate for WNV infection is 2% to 7%.2 Most of WNV-related mortalities occur in patients with neuroinvasive disease, where the mortality rate is 9%.2 In patients with WNV encephalitis, the mortality rate ranges from 12% to 15%, with the elderly having a much higher mortality rate, reaching as high as 35%.2 Most patients infected with WNV will be asymptomatic. Those who present with mild disease will usually recover in several days; patients suffering from more severe illness will have fatigue, muscle weakness, and aches that can persist for months.9 Most patients with WNV meningitis will make a full recovery as long as there are no associated focal neurologic deficits.2,9
The long-term outcomes for WNV meningo-encephaltitis vary, although patients with encephalitis are more likely to have a poorer outcome than those with just meningitis; symptoms such as fatigue, myalgia, residual tremor, and parkinsonism can persist for months to years, and in some cases, lifelong.23 Patients who experience AFP have the worst overall prognosis and usually will have significant residual weakness.2
Treatment
Currently, there is no specific treatment for WNV. General approaches to manage the disease involve the offering of supportive and symptomatic therapy. Supportive measures include respiratory support, fluid replacement for dehydration associated with nausea and vomiting, and prevention of secondary infection.2,18 Agents such as antipyretics and analgesics are commonly used to treat the symptoms of WNV.6 Short-course corticosteroids can be used to reduce cerebral edema, commonly seen in WNE, but the potential benefits of this treatment must be weighed against the possibility of potentiating the viral infection.19
Antiviral agents have been studied in WNV-infected cells in vitro or in laboratory animals or have been administered empirically to patients with WNE; these agents include purine and pyrimidine analogues, interferon (INF)-alfa, and human immunoglobulin.19 Data suggest that high concentrations of ribavirin can inhibit replication and cytopathogenicity of WNV in vitro, but its clinical efficacy is limited due to lack of controlled trials, ribavirin’s lack of effectiveness crossing the blood-brain barrier, and the challenge of identifying WNV early enough to improve outcomes.19,22 In addition, a retrospective Israeli study demonstrated no clinical benefit when oral ribavirin was administered empirically to patients during a WNV outbreak.27
INF-alfa, effective against hepatitis C, has shown to protect the spinal cord cells from becoming infected with WNV when given prior to exposure in vitro. Other in vitro studies involving monkey kidney cells have shown interferon’s ability to lengthen the survival of these cells when administered before or after WNV inoculation; these levels could be readily achieved in humans.19 While these results are promising, they have yet to be replicated in human studies; data from an open-label, nonblinded trial has suggested no clear benefit to use of INF-alfa.24
Another potential treatment option is intravenous immunoglobulin (IVIG). Animal models suggest rapid improvements in the course of the viral infection following administration of IVIG.22 This therapy may be an effective treatment for WNE, especially in immunocompromised patients; however, as with the previously mentioned treatments, no human studies have been done to prove the efficacy of IVIG.22
Ultimately, even if clinical data become available proving the efficacy of these or other agents in treating WNE, the challenge remains to deploy these agents in a timely manner postexposure to improve outcomes. Thus, improvements in the rapidity of diagnosis of WNV would be beneficial. Until then, supportive care is the basis of active treatment of WNV.
Prevention
Until effective treatment is available, prevention is the best way to manage WNV infection. Prevention focuses on education, control, and reduction of the mosquito population, elimination of breeding sites, and prevention of mosquito bites.22 The best plan is to avoid mosquito bites. The CDC promotes the use of insect repellants containing DEET (N,N-diethyl-meta-toluamide), the use of protective clothing such as long sleeves and pants, staying indoors between dusk and dawn when the mosquitoes responsible for spreading WNV are most active, draining standing water to eliminate breeding sites, placing mosquito netting over infant carriers when outdoors with infants, and installing and repairing screens.
The most effective insect repellents contain DEET. Different concentrations of DEET do not work better—they just provide longer protection. DEET is not recommended for children under the age of 2 months. For all other children, the American Academy of Pediatrics recommends using a product that contains a DEET concentration between 10% and 30%. When outdoors, protective clothing should be worn and sprayed with repellents since mosquitoes can bite through clothing.
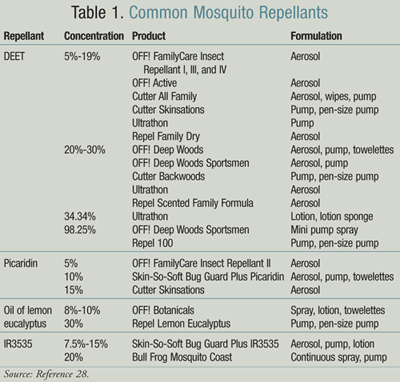
Other insect repellants approved by the CDC include picaridin, oil of lemon eucalyptus or PMD, and IR3535. The Environmental Protection Agency categorizes DEET and picaridin as “conventional repellants,” while oil of lemon eucalyptus, IR3535, and citronella are considered “biopesticide repellants,” which are derived from natural products (TABLE 1).28 The CDC added picaridin and oil of lemon eucalyptus to its approved insect repellant list in 2005. Picaridin is a repellant derived from peppers that is cosmetically pleasant and has low potential for toxicity, with effects lasting 2 to 8 hours. Oil of lemon eucalyptus is the most effective natural mosquito repellant. Its efficacy has been compared to lower concentrations of DEET with a shorter duration. IR3535 has been shown to be less effective than DEET and demonstrates variable protection time depending on testing methods utilized and mosquito species. Reported protection times varied from 6 minutes to 10 hours.28
People who use more than two protective measures can reduce their risk of WNV infection by 50%.29 Another preventive measure includes reporting dead birds to local authorities; this may be an indication that the WNV is circulating between birds and mosquitoes in the area. Local authorities should also be alerted to any standing water in places that could potentially serve as mosquito-breeding sites, such as storm sewers, ditches, and abandoned properties.18 Many state and local government agencies have implemented mosquito-control measures to protect the public health from mosquito-borne disease. For example, many counties in New York conduct mosquito sprayings to help reduce the mosquito population and risk of WNV.30,31 Other measures include mosquito surveillance, testing activities, and public education.
Conclusion
Since 1999, WNV has spread coast to coast in the U.S., and it will continue to be a public health concern for the future. Currently, prevention and control of WNV and mosquitoes are the only effective measures to prevent mortality associated with WNV infection. Pharmacists are in an ideal position to provide education to patients about WNV and its prevention. They are also able to assist patients in the selection and effective use of the most appropriate insect repellant.
REFERENCES
1. Murray KO, Mertens E, Despres P. West Nile virus and its emergence in the United States of America. Vet Res. 2010;41:67.
2. DeBiasi RL, Tyler KL. West Nile virus meningoencephalitis. Nat Clin Pract Neurol. 2006;2:264-275.
3. Petersen LR, Roehrig JT. West Nile virus: a reemerging global pathogen. Emerg Infect Dis. 2001;7:611-614.
4. Petersen LR, Marfin AA. West Nile virus: a primer for the clinician. Ann Intern Med. 2002;137:173-179.
5. Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
6. Azad H, Thomas S. West Nile encephalitis. Hosp Physician. 2004;40:12-16.
7. Petersen LR, Hayes EB. West Nile virus in the Americas. Med Clin North Am. 2008;92:1307-1322.
8. Hayes EB, O’Leary DR. West Nile virus infection: a pediatric perspective. Pediatrics. 2004;113:1375-1381.
9. Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. 2006;57:181-194.
10. Hayes EB, Komar N, Nasci RS, et al. Epidemiology and transmission dynamics of West Nile virus disease. Emerg Infect Dis. 2005;11:1167-1173.
11. Granwehr BP, Lillibridge KM, Higgs S, et al. West Nile virus: where are we now? Lancet Infect Dis. 2004;4:547-556.
12. CDC. Division of Vector Born Diseases. West Nile
virus. Entomology. April 2009.
www.cdc.gov/ncidod/dvbid/westnile/mosquitospecies.htm. Accessed May 13,
2012.
13. Mostashari F, Bunning ML, Kitsutani PT, et al.
Epidemic West Nile encephalitis, New York, 1999: results of a
household-based seroepidemiological survey. Lancet. 2001;358:261-264.
14. Pealer LN, Marfin AA, Petersen LR, et al. Transmission
of West Nile virus through blood transfusion in the United States in
2002. N Engl J Med. 2003;349:1236-1245.
15. O’Leary DR, Marfin AA, Montgomery SP, et al. The epidemic of West Nile virus in the United States, 2002. Vector Borne Zoonotic Dis. 2004;4:61-70.
16. Kumar D, Prasad GV, Zaltzman J, et al. Community-acquired West Nile virus infection in solid-organ transplant recipients. Transplantation. 2004;77:399-402.
17. Han LL, Popovici F, Alexander JP, et al. Risk factors for West Nile virus infection and meningoencephalitis, Romania, 1996. J Infect Dis. 1999;179:230-233.
18. Guharoy R, Gilroy SA, Noviasky JA, Ference J. West Nile virus infection. Am J Health Syst Pharm. 2004;61:1235-1241.
19. Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ. West Nile virus. Lancet Infect Dis. 2002;2:519-529.
20. Watson JT, Pertel PE, Jones RC, et al. Clinical characteristics and functional outcomes of West Nile fever. Ann Intern Med. 2004;141:360-265.
21. Petersen LR, Marfin AA, Gubler DJ. West Nile virus. JAMA. 2003;290:524-528.
22. Murray KO, Walker C, Gould E. The virology,
epidemiology, and clinical impact of West Nile virus: a decade of
advancements in research since its introduction into the Western
Hemisphere. Epidemiol Infect. 2011;139:810-817.
23. Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511-515.
24. Sejvar JJ, Marfin AA. Manifestations of West Nile neuroinvasive disease. Rev Med Virol. 2006;16:209-224.
25. Saad M, Youssef S, Kirschke D, et al. Acute flaccid
paralysis: the spectrum of a newly recognized complication of West Nile
virus infection. J Infect. 2005;51:120-127.
26. Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286-300.
27. Chowers MY, Lang R, Nassar F, et al. Clinical characteristics of the West Nile fever outbreak, Israel, 2000. Emerg Infect Dis. 2001;7:675-678.
28. Fradin MS. Insect repellants. Medscape Reference. January 2012. http://emedicine.medscape.com/article/1088411-overview#a1. Accessed July 6, 2012.
29. Loeb M, Elliott SJ, Gibson B, et al. Protective barrier and West Nile virus risk. Emerg Infect Dis. 2005;11:1433-1436.
30. Environmental Protection Agency. Pesticides: mosquito
control. May 2012. www.epa.gov/pesticides/health/mosquitoes/. Accessed
July 6, 2012.
31. Environmental Protection Agency. West nile virus:
region 2. October 2012. www.epa.gov/region02/pesticides/west_nile.htm.
Accessed July 6, 2012.
To comment on this article, contact rdavidson@uspharmacist.com.


