US Pharm. 2015;40(10):HS17-HS21.
ABSTRACT: Hereditary angioedema (HAE) is a rare genetic disease causing repeated swellings that can be life-threatening. HAE is caused by excessive bradykinin and neurokinin due to a deficient or defective serine protease inhibitor, plasma-derived C1 esterase inhibitor (pdC1-INH). Pharmacotherapy has significantly improved with the development of medications targeting the kinin pathway to prevent and reduce the duration and severity of attacks. Pharmacists’ understanding of these new drugs will ensure that medications are appropriately used and patient outcomes improved.
Hereditary angioedema (HAE) is a rare autosomal dominant disorder associated with inherited (75%) or spontaneous (25%) mutations of the HAE C1-INH (hereditary angioedema C1 esterase inhibitor) gene located on chromosome 11q12-q13.1.1,2 The result is recurrent nonpruritic edema of the skin and SC and submucosal tissues mainly occurring in the gastrointestinal (GI) and respiratory tracts, oropharynx, larynx, face, trunk, and genitalia.3,4
These episodes range from benign to fatal, manifesting as cosmetic disfigurement (swollen face), pain, nausea, vomiting, diarrhea, GI edema and bowel obstruction requiring hospitalization, and laryngeal swelling (<1% of attacks) potentially causing asphyxiation.5-7 The face and limbs are the most commonly affected, and swelling can be unilateral or bilateral. In some instances, prodromal symptoms may occur 3 to 24 hours before an attack, including nausea, fatigue, irritability, or erythema marginatum (a nonpruritic rash with ringlike lesions and sharp edges).8-10
Symptoms of the disease can occur annually or several times weekly and are typically self-limiting, generally resolving within 72 hours but potentially lasting up to 5 days until complement C4 is depleted.11 C4 is the factor activated by C1 esterase during classic pathway activation of the complement cascade. Unlike angioedema associated with histamine or immunoglobulin E (IgE) mediators, HAE is not adequately relieved by corticosteroids, antihistamines, or epinephrine.12 Instead, HAE requires treatments targeting bradykinin, the vasoactive molecule responsible for acute attacks.13
The early 2000s heralded advancements in FDA-approved medications used to treat and prevent HAE.14 New therapies can effectively relieve HAE symptoms and may prevent narcotics dependence caused by extreme pain and other psychological complications. This review introduces the new pharmacologic therapies available for the prevention and treatment of HAE.
Background and Pathophysiology
Angioedema has a number of etiologies categorized as acquired, allergic, drug-induced, or hereditary, among others. This review only covers HAE, a relatively rare disease with an incidence of 1:50,000 occurring in ethnicities and genders equally, with a mean age of onset of 11 years.2,5 Up to 50% of patients will develop at least one laryngeal attack in their lifetime.5
Hereditary angioedema is partitioned into types I, II, or III. Clinical presentation is similar among all types, and measurement of biomarkers is required to diagnose.15 Type I angioedema is caused by low serum concentrations of C1 esterase inhibitor (C1-INH) and represents 85% of cases, while type II is caused by normal or elevated concentrations of dysfunctional C1-INH protein in 15% of cases.16 C1-INH inhibits C1r and C1s in the complement pathway; factor XII, factor XI, and thrombin in the coagulation cascade; and kallikrein, the enzyme that cleaves kininogen to bradykinin.14 Ultimately, C1-INH deficiency or dysfunction leads to excessive bradykinin and neurokinin production.14 Bradykinin is the main mediator of HAE and causes the vasodilation and vascular permeability underlying attacks.14 Finally, type III HAE is the most rare, occurs mainly in women, and is associated with a mutation in coagulation factor XII in some patients.3,11 Factor XII activates kallikrein, leading to a similar end result of types I and II HAE bradykinin overproduction.13
General Principles of Management
Guidelines and recommendations for the management of HAE from four organizations—the World Allergy Organization (WAO) HAE International Alliance (2012)13; the Canadian Hereditary Angioedema Network (CHAEN), University of Calgary, and the Canadian Society of Allergy and Clinical Immunology (2010)15; and the Hereditary Angioedema International Working Group (2014)3—emphasize the following:
• Risk Factors: Certain drugs, such as ACE-inhibitors, hormone replacement therapy, and estrogen-based contraceptives, may provoke or exacerbate an attack and should be avoided.13,15 Nonpharmacologic risk factors include stress, trauma, infection, and pregnancy or menstruation.17,18
• Vaccinations: All patients should receive the hepatitis B vaccination when HAE is diagnosed, as they may require blood products for rescue therapy.15
• Medication Administration: Regardless of site, all attacks are eligible for treatment, and attacks affecting the airway must be treated.15 After an attack, patients should record the details of the event (i.e., possible precipitating factors, dose and medication administered, time to symptom relief, and adverse reactions).4
• Endpoints of Therapy: Pharmacologic agents are used to decrease the attack rate, hasten symptom relief, decrease symptom severity, and improve morbidity and mortality.3 Normalizing biomarkers of the complement pathways (C4 and C1-INH) should not be goals of therapy.19
Medications Used for the Prevention of HAE
There are three classes of medication used to prevent HAE episodes, including attenuated androgens, antifibrinolytics (tranexamic acid), and plasma-derived C1 esterase inhibitors (pdC1-INHs) (TABLE 1).15 Danazol and stanozolol (no longer available but possibly better tolerated) are attenuated androgens historically used and considered effective in decreasing severity and frequency of attacks.20-23 Long-term androgen therapy causes many adverse effects (e.g., weight gain, hepatic adenoma, emotional lability, vasomotor symptoms).24 Tranexamic acid is considered safer than androgens but is significantly less efficacious and generally not used in HAE prevention.25
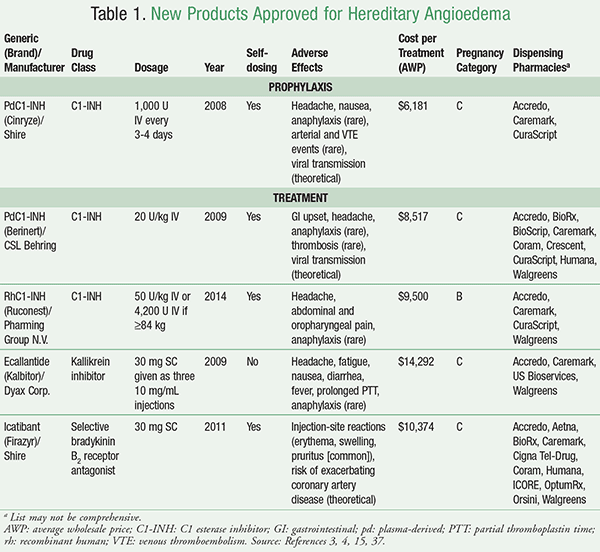
PdC1-INH is a product obtained from fractionated human plasma and marketed in the United States as Cinryze (indicated for routine prophylaxis of HAE) and Berinert (indicated for treatment of HAE). A third product (Cetor) is available in Europe.15 These IV agents supplement plasma with exogenous, functional C1-INH. Berinert and Cinryze are considered equally efficacious and safe, but given the cost difference between the two agents, some physicians have chosen to use Berinert off-label for prophylaxis.13 In a crossover trial, twice weekly pdC1-INH prophylaxis improved the 12-week attack rate, severity, and duration.26 Tachyphylaxis does not seem to occur with pdC1-INH prophylaxis, although some patients have required dose titration with prolonged use.27
As blood products, pdC1-INHs carry the theoretical risk of viral transmission, although there have been no documented cases since recent preparations have been brought to the U.S. market in 2008.15 Limited drug interactions and few adverse events have been associated with the use of pdC1-INHs.14 Ports for facilitating pdC1-INH administration are generally not recommended due to the increased risk of infection and thrombosis.4
Considerations in HAE Prevention
Short-term prophylaxis is used mainly in preprocedural scenarios and is favored for invasive or major surgeries, higher-risk procedures, surgical sites in close proximity to the respiratory tract, and procedures involving airway manipulation, or before situations that previously provoked an attack.13 However, minor procedures can also trigger attacks.13 For all procedures, two doses of on-demand therapy (Berinert) should be available in case symptoms recur or persist beyond the first dose.15 No comparative trials compare androgens against pdC1-INHs in short-term prophylaxis, but some prescribers may opt for Cinryze for its quick onset and robust half-life.3
Patients may qualify for long-term prevention if they cannot achieve satisfactory control or improvement in quality of life with on-demand therapy alone; have newly diagnosed severe disease; or have frequent airway attacks, limited access to acute care, comorbidities, or other complicating factors.28 Notably, attack frequency does not reliably indicate subsequent attack severity or location; as symptoms are always in flux, the need for long-term therapy should be routinely reevaluated.15
Medications Used in the Treatment of HAE Swellings
PdC1-INH (Berinert): Berinert is the orphan pdC1-INH approved for treatment of abdominal, facial, or laryngeal HAE attacks.29 Berinert relieves symptoms from an established attack significantly sooner than placebo when a weight-based dose (20 U/kg) is used.30 Patients have reported rash, headache, GI pain, nausea, muscle spasm, diarrhea, and vomiting after treatment, but these reactions are likely a result of the attack itself rather than medication side effects.30 SC Berinert for the treatment of HAE is being studied and may be available in 2 to 3 years.
RhC1-INH (Ruconest): The recombinant human C1 esterase inhibitor (rhC1-INH), Ruconest, has been effective in two placebo-controlled studies in reducing time to symptom relief relative to placebo.31 Ruconest is manufactured from the milk of transgenic rabbits, and patients with rabbit allergies have experienced anaphylactic reactions but human viral transmission is avoided.32 All patients must complete skin-prick or IgE serology testing to rabbit epithelium before commencing therapy.31 It has been proposed that allergy testing be repeated annually or after 10 treatments, whichever happens sooner.13 Ruconest is well tolerated; headaches are the most prevalent adverse effect but are likely disease-induced versus medication-induced.31
[Editor's note (10/23/15): These testing requirements only apply to the European labeling. In the U.S., the FDA-approved labeling does not require any testing.]
Ecallantide (Kalbitor): The kallikrein inhibitor ecallantide is supplied as three 10 mg SC injections to be administered consecutively as one dose.15 Post hoc analyses of two phase III controlled trials show that ecallantide treatment significantly improves response compared to placebo.33 Hypersensitivity reactions are the greatest risk of treatment, and an estimated 1.6% of patients have experienced anaphylaxis.34 As a result, ecallantide should only be administered by a healthcare professional.24
Icatibant (Firazyr): Icatibant is a selective, competitive antagonist at the bradykinin B2 receptors that has reduced time to significant symptom relief faster than tranexamic acid.15,35 There have been no reported allergic reactions to icatibant, but transient local injection-site reactions occur in most patients (~97%).36 Since anaphylaxis has not been observed, self-dosing is permitted.15
TABLE 1 provides additional information regarding pharmacologic treatments for HAE.3,4,15,37
Considerations for HAE Treatment
Drugs used for the treatment of HAE attacks directly or indirectly antagonize bradykinin to disrupt the propagation of the complement cascade. Treatment is appropriate for all attacks, and only with cutaneous attacks at sites other than the face or neck should patients withhold treatment if desired.15 All drugs are effective for treating attacks occurring in any region of the body.10 Inconsistencies in study design do not allow for reliable comparisons of on-demand therapies.38 Relief from treatment typically takes 30 to 60 minutes, and generally anywhere from 10% to 30% of attacks require a second dose.4
Before the introduction of new agents, fresh frozen plasma (FFP) was frequently used to treat attacks. FFP may be effective as a treatment option but contains substrates of C1-INH that may worsen an attack and should be reserved for last-line therapy.15
Role of the Pharmacist
Most healthcare professionals have had limited experience or exposure with new orphan products. As a result, pharmacists are increasingly responsible for understanding how to obtain products from specialty pharmacies, educating patients on appropriate administration, and ensuring that medication (e.g., Berinert) is stored in emergency departments for attack treatment. These new medications are extremely costly, with estimates based upon average wholesale price (AWP) suggesting up to $500,000 or more per year. A recent analysis indicates that the costs for HAE therapy have tripled in the last 2 years and that specialty drug treatment costs are on average more than $300,000 annually.39 Each manufacturer has a financial assistance program that pharmacists should work with to obtain income-based coupons for indigent populations.
Given that these drugs work best when administered as soon as possible, pharmacists must advocate to the healthcare team and patients the benefits of self-administration.7 Before coaching patients, pharmacists should review the manufacturers’ websites for relevant dosing/infusing calculators and instructions for preparing, storing, and administrating the medications. Pharmacists and nurses employed by the manufacturers may also be available for instruction on product administration.
Summary
Recent advancements have been made with medications used to prevent and treat HAE. Given the paucity of evidence supporting definitively preferred prevention or treatment regimens, the pharmacist is relied upon for understanding the interplay of medication risk and benefit profiles with patient circumstances. It is important for pharmacists, as part of the healthcare team, to be up-to-date on these new treatments in order to optimize therapy in patients with HAE.
REFERENCES
1. Bock SC, Skriver K, Nielsen E, et al. Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry. 1986;25:4291-4301.
2. Zuraw BL. Clincal practice: hereditary angioedema. N Engl J Med. 2008;359:1027-1036.
3. Cicardi M, Aberer W, Banerji A, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602-616.
4. Riedl M. Creating a comprehensive treatment plan for hereditary angioedema. Immunol Allergy Clin North Am. 2013;33(4):471-485.
5. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119:267-274.
6. Bork K, Staubach P, Eckardt AJ, Hardt J. Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol. 2006;101:619-627.
7. Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474-481.
8. Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med. 1976;84:586-593.
9. Prematta MJ, Kemp JG, Gibbs JG, et al. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc. 2009;30:506-511.
10. Burke JB. Erythema marginatum. Arch Dis Child. 1955;30:359-365.
11. Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33(suppl 1):S145-S156.
12. Frank MM. Hereditary angioedema. J Allergy Clin Immunol. 2008;121(suppl):S398-S401.
13. Craig T, Aygören-Pürsün E, Maurer M, et al. WAO guideline for the management of hereditary angioedema. World Allergy Organ J. 2012;5(12):182-199.
14. Bork K. An evidence-based therapeutic approach to hereditary and acquired angioedema. Curr Opin Allergy Clin Immunol. 2014;14(4):354-362.
15. Bowen T, Cicardi M, Farkas H, et al. 2010 international consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6(1):24.
16. Agostoni A, Aygören-Pürsün E, Binkley KE, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114(suppl):S51-S131.
17. Zotter Z, Csuka D, Szabó E, et al. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. 2014;9:44.
18. Morcavallo PS, Leonida A, Rossi G, et al. Hereditary angioedema in oral surgery: overview of the clinical picture and report of a case. J Oral Maxillofac Surg. 2010;68:2307-2311.
19. Warin A, Greaves M, Gatecliff M, et al. Treatment of hereditary angioedema by low dose attenuated androgens: disassociation of clinical response from levels of C1 esterase inhibitor and C4. Br J Dermatol. 1980;103:405-409.
20. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
21. Cicardi M, Castelli R, Zingale LC, Agostoni A. Side effects of long-term prophylaxis with attenuated androgens in hereditary angioedema: comparison of treated and untreated patients. J Allergy Clin Immunol. 1997;99:194-196.
22. Sloane DE, Lee CW, Sheffer AL. Hereditary angioedema: safety of long-term stanozolol therapy. J Allergy Clin Immunol. 2007;120:654-658.
23. Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005;139:379-394.
24. Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67(2):147-157.
25. Sheffer AL, Austen KF, Rosen FS. Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med. 1972;287:452-454.
26. Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
27. Bork K, Hardt J. Hereditary angioedema: long-term treatment with one or more injections of C1 inhibitor concentrate per week. Int Arch Allergy Immunol. 2011;154:81-88.
28. Lang D, Aberer W, Zuraw B, et al. International consensus on hereditary and acquired angioedema. Ann Allergy Asthma Immunol. 2012;109(6):395-402.
29. Berinert (C1 esterase inhibitor, human) package insert. Marburg, Germany: CSL Behring; 2015.
30. Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. 2009;124:801-808.
31. Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. 2010;126:821-827.
32. Longhurst H. Rhucin, a recombinant C1 inhibitor for the treatment of hereditary angioedema and cerebral ischemia. Curr Opin Investig Drugs. 2008;9: 310-323.
33. Banta E, Horn P, Craig TJ. Response to ecallantide treatment of acute attacks of hereditary angioedema based on time to intervention: results from the EDEMA clinical trials. Allergy Asthma Proc. 2011;32:319-324.
34. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
35. Maurer M, Aberer W, Bouillet L, et al. Hereditary angioedema attacks resolve faster and are shorter after early icatibant treatment. PLoS One. 2013;8:e53773.
36. Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532-541.
37. Bork K. An evidence based therapeutic approach to hereditary and acquired angioedema. Curr Opin Allergy Clin Immunol. 2014;14(4):354-362.
38. Parikh N, Riedl MA. New therapeutics in C1INH deficiency: a review of recent studies and advances. Curr Allergy Asthma Rep. 2011;11(4):300-308.
39. Poquette C, Starner CI, Hall S, Gleason PP. Hereditary angioedema drug utilization and spend: a medical and pharmacy integrated analysis. JMCP. 2015;21(suppl 4a):S16.
To comment on this article, contact rdavidson@uspharmacist.com.



