US Pharm. 32(11)HS16-HS32
Drug-induced movement disorders (DIMDs), also commonly referred to as
extrapyramidal symptoms (EPS), represent a variety of iatrogenic and
clinically distinct movement disorders, including akathisia, tardive
dyskinesia, dystonia, and parkinsonism
(
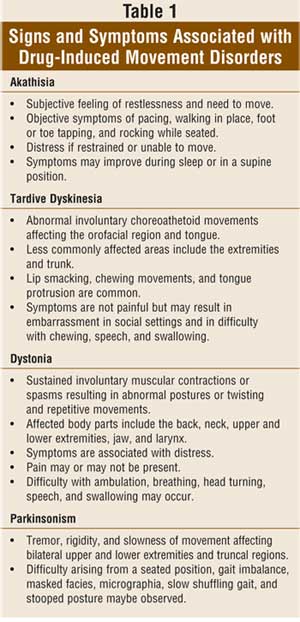
TABLE 1
)
. DIMDs remain a significant burden among certain patient populations, such as
those receiving treatment with dopamine receptorÒblocking agents (DRBAs; e.g.,
various psychotropic agents and antiemetics)
(
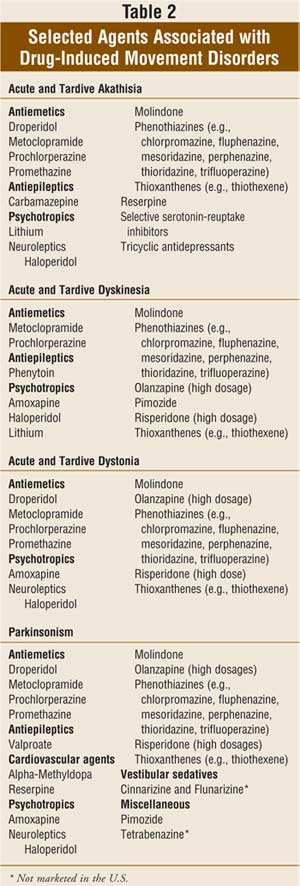
TABLE 2
)
.
DIMDs are often underrecognized, and knowledge of DIMDs will allow clinicians,
pharmacists, and other health care professionals to better identify and manage
patients with these conditions.
Symptoms of DIMDs interfere with social functioning, interpersonal
communication, and performance of motor tasks and activities of daily living.
In addition to reduced quality of life, patients experiencing DIMDs tend to
abandon therapy (which may result in disease relapse and rehospitalization).
The onset of a DIMD can be classified as acute (i.e., occurring within hours
to days after exposure), subacute (i.e., occurring weeks after drug exposure),
and tardive (i.e., occurring months to years after drug exposure). The
symptomatology of DIMDs (
TABLE 1
) is often indistinguishable from that of idiopathic movement disorders and
may resemble that of several other medical conditions. However, with a careful
drug history, recognition of a DIMD may be relatively straightforward, as in
the case of a patient with a history of DRBA treatment presenting with classic
symptoms of tardive dyskinesia (TDk). As with idiopathic movement disorders,
anxiety and stress will also exacerbate symptoms associated with DIMDs. In
general, elderly females are more susceptible to develop TDk, young males more
susceptible to dystonic reactions, and the elderly more prone to drug-induced
parkinsonism. Also notable is that patients may develop mixed DIMDs (i.e.,
they may have more than one type of DIMD).
Drugs
Agents implicated in DIMDs are listed in
TABLE 2.
The centrally acting DRBAs, such as haloperidol and phenothiazine
neuroleptics, are the agents most commonly associated with DIMDs. DIMDs are
less frequently associated with the atypical antipsychotics, but dose-related
EPS occurs with olanzapine and risperidone (especially at dosages greater than
6 mg/day).
Movement disorders are also associated with other medications, such as
antiemetics that block central dopamine receptors (i.e., droperidol,
metoclopramide, and prochlorperazine), lithium, selective serotonin reuptake
inhibitors (SSRIs), stimulants, and tricyclic antidepressants (TCAs). Tremor
commonly occurs with lithium treatment and occasionally chorea.1
SSRIs can commonly cause tremor and, less commonly, dyskinesia, dystonia, or
parkinsonism.2
Stimulant drugs (e.g., amphetamine, methylphenidate, and pemoline) have been
known to produce a variety of movement disorders such as dyskinesias,
dystonia, stereotypic behavior, and tics.3 The most common movement
disorders associated with TCAs are myoclonus and tremor.4 The
antiepileptic drug valproate is commonly associated with tremor. For many
years, chorea has been recognized as a complication of estrogen- and
progesterone-containing products.5 Psychotherapeutic combination
products containing a neuroleptic, such as perphenazine/amitriptyline, should
not be overlooked as causative agents.
Prevalence
Neuroleptic-induced movement disorder occurs in a significant number of
patients who are treated with conventional neuroleptics. In one study of
chronic institutionalized patients with schizophrenia, conventional
neuroleptics were associated with movement disorders in 61.6% of patients.
6 Of those patients, 31.3% had akathisia, 23.2% had parkinsonism, and
32.3% had TDk. However, with the advent of the atypical antipsychotics, rates
of neuroleptic-induced movement disorders may have declined. In another study
of 125 patients who developed movement disorders from DRBAs, 63% had tardive
dyskinesia, 30% had parkinsonism, 24% had dystonia, and 7% had akathisia.7
Haloperidol was implicated in 37% of cases, followed by the combination
amitriptyline with perphenazine in 30%, thioridazine in 27%, chlorpromazine in
20%, and metoclopramide in 8%.
AKATHISIA
The word
akathisia
is a derivative of the Greek word meaning "not to sit." Neuroleptic-induced
acute and tardive akathisia is a common and distressing adverse effect that is
associated with poor treatment adherence and, ultimately, an increased risk of
psychiatric relapse. Conventional neuroleptics and phenothiazine antiemetics
are most commonly implicated. Other drugs commonly associated with akathisia
are listed in
TABLE 2.
The atypical antipsychotics are associated with lower rates of akathisia,
with reported values approximately two to three times less than those of
conventional neuroleptics. In one study, the prevalence of akathisia
associated with clozapine, risperidone, and conventional neuroleptics was
7.3%, 13%, and 23.8%, respectively.8 Akathisia is also an adverse
effect of SSRIs and appears to occur in at least 4.5% of exposed patients.
9
Clinical
Features and Risk Factors:
Neuroleptic-induced akathisia is comprised of both a subjective and an
objective component (
TABLE 1
). Acute drug- induced akathisia should be suspected if symptoms developed
soon after initiation of implicated drugs and in the absence of other
conditions associated with restlessness. Symptoms of acute akathisia typically
occur within four weeks of initiating or increasing the dosage of the
offending drug and may also develop after neuroleptic cessation or dosage
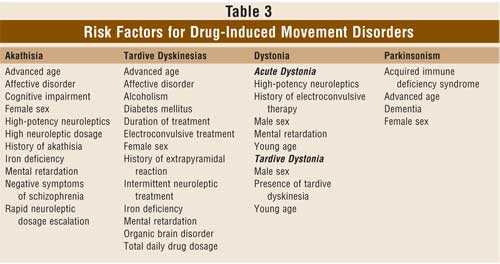
reduction (i.e., withdrawal akathisia). Risk factors for akathisia are listed
in
TABLE 3.10,11
The symptoms of drug-induced akathisia can be mistaken for other medical
conditions (e.g., restless legs syndrome), for agitation and anxiety
presenting as part of a psychiatric disorder, and for drug withdrawal
syndromes.
Therapeutic
Management:
When left untreated, the symptoms of acute akathisia may gradually subside or
may wax and wane over time. In some patients, acute akathisia may become
chronic and persistent for months or years. Acute akathisia has been
associated with promoting aggression, psychopathology, suicidal behavior, and
treatment nonadherence in patients with schizophrenia.12 Therefore,
prevention is a key component of treatment. Standardized titration to avoid
excessive dosage escalation and the use of atypical antipsychotics are
successful measures of prevention. Although iron deficiency has been
associated with akathisia, routine iron supplementation as a preventive or
treatment intervention is not supported by the available evidence.13
It is reasonable to correct underlying iron deficiencies or to administer
oral iron supplementation in akathisia unresponsive to standard measures. In a
patient at high risk for akathisia, prophylactic treatment with an
antimuscarinic agent or beta-blocker is reasonable.
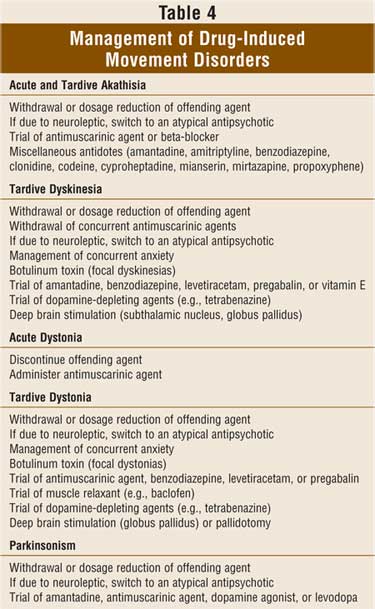
The management of akathisia is summarized in
TABLE 4.
In the patient experiencing acute akathisia, the causative agent should be
discontinued, if possible. A switch to an atypical antipsychotic or
alternative agent should be considered. Administration of a lipophilic
beta-blocker, such as propranolol, is effective and well tolerated.14
Beta2-receptor blockade appears to be crucial for efficacy as beta
1-receptor selective agents are less effective.15 However, a
trial of a cardioselective, beta1-receptor blocker is recommended
if a nonselective beta-blocker is contraindicated or is not tolerated. The
hydrophilic beta-blockers (e.g., atenolol and nadolol) do not appear to be
effective. Administration of antimuscarinic agents (e.g., benztropine,
diphenhydramine), benzodiazepines, or antiserotonergic agents (cyproheptadine)
are also effective and may be preferred if sedation is desired.16-18
TARDIVE DYSKINESIA
Since it was first reported in the United States in 1960, TDk has become the most recognized movement disorder induced by neuroleptic treatment.19 Tardive dyskinesia develops after at least one month's exposure to DRBAs, and, if the offending drug is continued, remission is rare. Drugs that are associated with TDk are listed in TABLE 2. The term tardive emphasizes the delayed onset of involuntary, choreiform, orofacial movements secondary to neuroleptic use. Although symptoms may initially be mild, many patients develop progressively severe TDk, resulting in meaningful disability and disfigurement. Use of high-dose neuroleptics and longer duration of treatment are associated with increased risk of developing TDk. In young patients, approximately 5% will develop TDk with each year of neuroleptic exposure.20 The elderly population appears to have the highest incidence and prevalence of TDk and is at least five times more at risk for developing TDk as compared to young patients. 21 However, with the use of the newer atypical antipsychotics, the risk of developing TDk is reduced.22 In one review of 11 studies, atypical antipsychotics were associated with a mean annual incidence of new-onset tardive dyskinesia of 0.8% in the adults as compared to 5.4% in adults treated with haloperidol.22 These data suggest that the risk of TDk associated with atypical antipsychotics is at least one-fifth that of conventional neuroleptics. In general, if patients do not develop TDk during the initial five years of neuroleptic treatment, the risk of developing TDk during later years is reduced.
Clinical Features and Risk
Factors: Signs and
symptoms of TDk are listed in TABLE 1. Frequently, TDk may occur in the
presence of other movement disorders, such as akathisia, dystonia, or
parkinsonism. The onset of TDk is insidious and symptoms are initially mild.
Often, the symptoms are unnoticeable to patients but are more apparent to
family members or bystanders. Even mild cases of TDk are associated with
social impairment such as employment difficulties, social isolation, and
stigma. In more severe cases, functional impairment occurs. Patients
experience difficulties with chewing, speaking, and swallowing. Orofacial
dyskinesias may also result in dental problems, denture displacement, and
damage to the soft tissues within the oral cavity.
When assessing for presence
and severity of TDk, the Abnormal Involuntary Movements Scale (AIMS),
developed by the National Institute of Mental Health, is a commonly employed
instrument, particularly in the psychiatric field.23 The AIMS rates
dyskinetic movements in seven body regions and includes assessments for global
severity, functional impairment, and self-awareness of symptoms. Assessment
tools, such as the AIMS, may be utilized every three months to reassess
specific symptoms of TDk. Factors associated with exacerbation of TDk symptoms
include administration of antimuscarinics or sympathomimetic stimulants and
emotional extremes. As with most dyskinesias, symptoms subside during sleep
and, in mild cases, patients may be unaware of the movements.
Age is a well-established risk
factor for TDk. Other risk factors are listed in TABLE 3. Not only is
TDk more common in the elderly, but the condition tends to be more severe and
irreversible in this age group. If multiple risk factors are present, an
additive effect on risk potential is observed; for example, elderly females
are very susceptible to TDk.
Therapeutic Management:
Once TDk develops, remission rates are low if the antipsychotic is continued.
In one study, only 11% of patients improved over the course of five years.
24 Early detection of TDk is imperative, as remission rates are
inversely correlated with duration and severity of TDk. If detected early and
the offending agent is discontinued, TDk remission rates are favorable,
especially in the younger population, but may require several months to years
to resolve. However, the benefit of discontinuing the offending antipsychotic
for the purpose of treating TDk should be weighed against the likelihood of
psychotic decompensation. Additionally, patients with severe TDk and the
elderly are less likely to experience remission, even with elimination of the
offending agent. Occasionally, TDk may occur also after withdrawal of chronic
DRBA treatment (withdrawal TDk). This type of TDk is reported most commonly in
children and generally improves within three months.25
A summary of the management of
TDk is provided in TABLE 4. Treatment for TDk varies and includes drug
discontinuation, switching to or initiating an atypical antipsychotic,26
discontinuing anticholinergics (except in tardive dystonia), and initiating
adjunctive vitamin E or benzodiazepines. For patients with severe and
refractory TDk, neurosurgical treatments may be effective.27
Anxiety often exacerbates TDk and should be treated appropriately. In a recent
meta-analyses of evidence from randomized, controlled trials, more than 500
trials evaluating over 90 different interventions were identified.28
The analysis did not yield any definitive information on how best to treat
TDk. Given the lack of good evidence, emphasis is placed on primary
prevention, prompt recognition, and management of early and potentially
reversible causes. It is reasonable for clinicians to consider the use of
atypical antipsychotics for long-term therapy over that of the older
neuroleptics as a means to prevent or reduce the risk for TDk. Other
strategies include use of the lowest effective DRBA dosage and elimination
unnecessary, prolonged drug exposure. Although the use of intermittent
antipsychotic treatment (or drug holidays) may seem like a logical strategy
for reducing TDk risk, it is actually associated with an increased risk of TDk
and higher rates of psychosis relapse and rehospitalization.29 For
neuroleptic-induced TDk, withdrawal of the offending agent may result in
partial improvement in up to 50% of patients. But overall, complete and
long-lasting resolution of TDk is uncommon.30
DYSTONIA: ACUTE AND TARDIVE
Drugs associated
with dystonia are listed in TABLE 2. The development of drug-induced
acute dystonia (DIAD) is more commonly associated with butyrophenone
antipsychotics (e.g., haloperidol) as compared to phenothiazine antipsychotics
(e.g., chlorpromazine).31 Tardive dystonia, defined as an
involuntary movement predominated by dystonia and associated with the use of a
DRBA, is distinct from TDk, although both often coexist in the same patient.
32
Clinical Features and Risk
Factors: The onset of
DIAD is sudden, and most cases occur within hours to several days of initial
exposure to DRBAs or, less commonly, after a DRBA dosage increase or a
reduction in concomitant antimuscarinic agent. The occurrence of DIAD appears
to be greater in younger patients and in patients receiving parenteral DRBAs.
Signs and symptoms of dystonia are listed in TABLE 1. The severity of
symptoms and anatomical distribution vary, but the classic clinical
presentation is characterized by the "three O's": oculogyric crisis (conjugate
deviation of the eyes upward or laterally), opisthotonos (involuntary
posturing in which the head, neck, and spine are arched backward), and
oromandibular dystonia (causing difficulty in opening or closing the mouth).
Symptoms are usually painful and, in severe cases, DIAD may be life
threatening. Rhabdomyolysis due to sustained muscle contraction may also
occur. If dystonia occurs in the presence of fever, generalized rigidity,
altered level of consciousness, and autonomic instability, a diagnosis of
neuroleptic malignant syndrome should be considered. Owing to the discomfort
associated with DIAD, patients are at higher risk for abandoning medication
therapy, or nonadherence.
Risk factors for the
development of acute dystonia are listed in TABLE 3.33 As
with other DIMDs, the presence of multiple risk factors has an additive
effective on risk. An inverse relationship exists between the incidence of
DIAD and age. In contrast to TDk, dystonia is uncommon in older patients. The
highest risk population is young males on high-potency neuroleptics.
Unlike acute dystonia, tardive
dystonia develops insidiously after months to years of treatment with a DRBA
or within three months after treatment discontinuation and often coexists with
TDk. In tardive dystonia, remission is uncommon. Children are at particularly
greater risk of tardive dystonia compared to adults. Symptoms may or may not
be painful and can be isolated to one body part or may spread to contiguous
body parts (segmental dystonia), or even generalize to multiple body parts.
Other medical conditions that present with insidious development of dystonia
include idiopathic dystonia, Huntington's disease, Wilson's disease,
levodopa-responsive dystonia, and conversion reaction. In addition, other
conditions that cause a twisted neck, such as orthopedic or congenital
problems of the neck, ophthalmologic conditions resulting in head tilt to
compensate for vision problems, stiff neck, arthritis, or wry neck, should be
ruled out.
Therapeutic Management:
The management of acute and tardive dystonias is summarized in TABLE 4
. Several studies have reported that the administration of concurrent
antimuscarinic agents (e.g., benztropine, diphenhydramine, and
trihexyphenidyl) as a preventative measure reduces the overall rate of
dystonia by at least two-fold.31,34,35 In patients treated with
high-potency neuroleptics, the reduction is even greater at five- to 11-fold.
34,36 Since the greatest risk of neuroleptic-induced acute dystonia
occurs within the first week of drug treatment, the short-term administration
of oral antimuscarinic agents may be considered, especially in young patients
receiving high-potency antipsychotics. The efficacy of antimuscarinic
prophylaxis appears to be inversely related to age of the patient. Due to
reduced prophylactic efficacy and undesirable antimuscarinic adverse
reactions, the use of antimuscarinic agents in elderly patients for primary
prevention of DIAD is disfavored.
An acute dystonic episode can
be effectively relieved with a short course of a potent antimuscarinic agent
(e.g., benztropine, diphenhydramine) administered orally, intramuscularly, or
intravenously. If life-threatening dystonia is present, intravenous
administration is warranted and supportive measures are required.
Benzodiazepines may be administered if use of an antimuscarinic agent is
contraindicated.
In tardive dystonia, few
treatments have proven to be consistently useful, with the exception of
botulinum toxin. Therefore, prevention is important. The chronic use of
conventional DRBAs (e.g., neuroleptics and metoclopramide) should be carefully
evaluated and alternatives considered. For medical management, atypical
antipsychotics, benzodiazepines, muscle relaxants, and dopamine-depleting
drugs, such as tetrabenazine, may be effective and can be used in combination
with antimuscarinics. Less commonly used drugs include amantadine,
beta-blockers, clonidine, dantrolene, levodopa, and antiepileptics such as
levetiracetam, pregabalin, tiagabine, and zonisamide. For focal dystonic
symptoms, local injections of botulinum toxin are preferred due to relative
lack of systemic adverse effects and consistent efficacy among patients. In
refractory and severe cases, intrathecal baclofen or neurosurgical
interventions may be effective.37,38
PARKINSONISM
Drug-induced
parkinsonism (DIP) is considered the second most common form of parkinsonism.
Drugs that may induce or exacerbate parkinsonism are listed in TABLE 2.
Of all drugs, the DRBAs are most commonly implicated. In one study, the
prevalence of parkinsonian rigidity associated with clozapine, risperidone,
and conventional neuroleptics was 4.9%, 17.4%, and 35.7%, respectively.8
In addition, drugs that deplete presynaptic dopamine (e.g., reserpine,
tetrabenazine) or act as dopamine "false transmitters" (e.g.,
alpha-methyldopa) may also induce parkinsonism. Valproate is an
underrecognized source of DIP due to insidious onset of parkinsonian symptoms.
39 The SSRIs are uncommonly associated with DIP, although SSRI-induced
tremor is common.
Clinical Features and
Risk Factors: In
general, the clinical features are indistinguishable from idiopathic
Parkinson's disease and include at least two of the following: tremor (rest or
postural), rigidity, and bradykinesia (TABLE 1). In the absence of an
accurate drug history, symptoms of DIP can be easily mistaken for idiopathic
Parkinson's disease. The presence of DIP may be overlooked because symptoms
such as reduced facial expression, reduced energy and motivation, and
bradykinesia can mimic negative symptoms of schizophrenia as well as
psychomotor retardation associated with depressive disorders. In the majority
of cases, DIP is a subacute process that develops within three months of
initiating the offending agent or, in some cases, after a dosage increase, and
it is slowly reversible upon drug withdrawal. Risk factors for the development
of DIP are listed in TABLE 3. Patients at greatest risk are those who
are demented, elderly, or have preexisting Parkinson's disease. Also, patients
with acquired immune deficiency syndrome (AIDS) appear to be very susceptible
to DIP. In one study, the likelihood of developing EPS was 2.4 times higher
among patients with AIDS as compared to psychotic patients without AIDS.40
Therapeutic Management:
The management of DIP is summarized in TABLE 4. Upon drug withdrawal,
symptoms of DIP resolve slowly, over months to years. Other strategies
utilized in the management of DIP include dosage reduction, switching to an
atypical antipsychotic agent, and the use of amantadine and antimuscarinic
agents. The routine use of antimuscarinic agents for primary prophylaxis of
DIP is controversial, especially for elderly patients.
CONCLUSION
Drug-induced
movement disorders (i.e., akathisia, tardive dyskinesia, dystonia, and
parkinsonism) pose a significant burden to patients and may result in
medication nonadherence or abandonment of therapy. Dopamine-receptor blocking
agents such as conventional antipsychotics (e.g., haloperidol and
chlorpromazine) and antiemetics (e.g., metoclopramide and prochlorperazine)
are commonly implicated. Clinicians and pharmacists should recognized that
DIMDs can occur acutely (i.e., hours to days after drug exposure), subacutely
(i.e., within weeks after exposure), or months to years after drug exposure.
With the exception of drug-induced acute dystonia, the medical and surgical
treatment of most DIMDs does not consistently provide benefit; therefore,
prevention is essential. Knowledge of DIMDs should allow health care
professionals to better identify patients with DIMDs or those at risk for them
and implement treatment and prevention plans.
References
1. Gelenberg AJ, Jefferson JW. Lithium tremor. J Clin Psychiatry. 1995;56:283-287.
2. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother . 1998;32:692-698.
3. Weiner WJ, Sanchez-Ramos J. Movement disorders and dopamimetic stimulant drugs: In: Lang AE, Weiner WJ, eds. Drug-Induced Movement Disorders. Mount Kisco, NY: Futura Publishing Co.; 1992:315-337.
4. Vandel P, Bonin B, Leveque E, Sechter D, et al. Tricyclic antidepressant-induced extrapyramidal side effects. Eur Neuropsychopharmacol. 1997;7: 207-212.
5. Vela L, Sfakianakis GN, Heros D, et al. Chorea and contraceptives:
case report with pet study and review of the literature. Mov Disord. 2004;19:349-352.
6. Janno S, Holi M, Tuisku K, Wahlbeck K. Prevalence of neuroleptic-induced movement disorders in chronic schizophrenia inpatients. Am J Psychiatry. 2004;161:160-163.
7. Miller LG, Jankovic J. Neurologic approach to drug-induced movement disorders: a study of 125 patients. South Med J. 1990;83:525-532.
8. Miller CH, Mohr F, Umbricht D, et al. The prevalence of acute extrapyramidal signs and symptoms in patients treated with clozapine, risperidone, and conventional antipsychotics. J Clin Psychiatry. 1998;59:69-75.
9. Baldassano CF, Truman CJ, Nierenberg A, et al. Akathisia: a review and case report following paroxetine treatment. Compr Psychiatry. 1996;37:122-124.
10. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21:451-461.
11. Kuloglu M, Atmaca M, Ustundag B, et al. Serum iron levels in schizophrenic patients with or without akathisia. Eur Neuropsychopharmacol. 2003;13:67-71.
12. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review.
J Forensic Sci. 2003;48:187-189.
13. Gold R, Lenox RH. Is there a rationale for iron supplementation in the treatment of akathisia? A review of the evidence. J Clin Psychiatry. 1995; 56:476-483.
14. Fischel T, Hermesh H, Aizenberg D, et al. Cyproheptadine versus propranolol for the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. J Clin Psychopharmacol. 2001;21:612-615.
15. Zubenko GS, Lipinski JF, Cohen BM, et al. Comparison of metoprolol and propranolol in the treatment of akathisia. Psychiatry Res. 1984;11:143-149.
16. Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26:265-270.
17. Lima AR, Weiser KV, Bacaltchuk J, Barnes TR. Anticholinergics for neuroleptic-induced acute akathisia (Cochrane Review). In: The Cochrane Library, Issue 2, 2004. Chichester, UK: John Wiley & Sons, Ltd.
18. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia (Cochrane Review). In: The Cochrane Library, Issue 2, 2004. Chichester, UK: John Wiley & Sons, Ltd.
19. Kruse W. Persistent muscular restlessness after phenothiazine treatment: report of 3 cases. Am J Psychiatry. 1960;117:152-153.
20. Woerner MG, Kane JM, Lieberman JA, et al. The prevalence of tardive dyskinesia. J Clin Psychopharmacol . 1991;11:34-42.
21. Jeste DV. Tardive dyskinesia rates with atypical antipsychotics in older adults. J Clin Psychiatry. 2004;65(suppl 9):21-24.
22. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161:414-425.
23. Guy W. Abnormal Involuntary Movements Scale (AIMS). ECDEU Assessment Manual for Psychopharmacology. Washington, DC: United States Government Printing Office; 1976:534-537.
24. Bergen JA, Eyland EA, Campbell JA, et al. The course of tardive dyskinesia in patients on long-term neuroleptics. Br J Psychiatry. 1989;154:523-528.
25. Polizos P, Engelhardt DM. Dyskinetic phenomena in children treated with psychotropic medications. Psychopharmacol Bull. 1978;14:65-68.
26. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65:696-701.
27. Schrader C, Peschel T, Petermeyer M, et al. Unilateral deep brain stimulation of the internal globus pallidus alleviates tardive dyskinesia. Mov Disord. 2004;19:583-585.
28. Soares-Weiser K, Fernandez HH. Tardive dyskinesia. Semin Neurol. 2007;27:159-169.
29. van Harten PN, Hoek HW, Matroos GE, et al. Intermittent neuroleptic treatment and risk for tardive dyskinesia: Curacao Extrapyramidal Syndromes Study III. Am J Psychiatry. 1998;155:565-567.
30. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
31. Spina E, Sturiale V, Valvo S, et al. Prevalence of acute dystonic reactions associated with neuroleptic treatment with and without anticholinergic prophylaxis. Int Clin Psychopharmacol. 1993;8:21-24.
32. Burke RE, Fahn S, Jankovic J, et al. Tardive dystonia: late-onset and persistent dystonia caused by antipsychotic drugs. Neurology. 1982;32:1335-1346.
33. Ballerini M, Bellini S, Niccolai C, et al. Neuroleptic-induced dystonia: incidence and risk factors. Eur Psychiatry. 2002;17:366-368.
34. Arana GW, Goff DC, Baldessarini RJ, et al. Efficacy of anticholinergic prophylaxis for neuroleptic-induced acute dystonia. Am J Psychiatry. 1988;145:993-996.
35. Goff DC, Arana GW, Greenblatt DJ, et al. The effect of benztropine on haloperidol-induced dystonia, clinical efficacy and pharmacokinetics: a prospective, double-blind trial. J Clin Psychopharmacol. 1991;11:106-112.
36. Boyer WF, Bakalar NH, Lake CR. Anticholinergic prophylaxis of acute haloperidol-induced acute dystonic reactions. J Clin Psychopharmacol. 1987;7:164-166.
37. Dressler D, Oeljeschlager RO, Ruther E. Severe tardive dystonia: treatment with continuous intrathecal baclofen administration. Mov Disord. 1997;12:585-587.
38. Kupsch A, Kuehn A, Klaffke S, et al. Deep brain stimulation in dystonia.
J Neurol. 2003;250(suppl 1):I47-I52.
39. Jamora D, Lim SH, Pan A, et al. Valproate-induced Parkinsonism in epilepsy patients. Mov Disord. 2007;22:130-133.
40. Hriso E, Kuhn T, Masdeu JC, et al. Extrapyramidal symptoms due to dopamine-blocking agents in patients with AIDS encephalopathy. Am J Psychiatry. 1991;148:1558-1561.
To comment on this article, contact editor@uspharmacist.com.


