US Pharm. 2018;(43):42-47.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2017–2018 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Ertugliflozin (Steglatro, Merck)
Indication and Clinical Profile1,2: Ertugliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor that has been approved for the treatment of type 2 diabetes (DM2) in adult patients as an adjunct to diet and exercise. DM2, a chronic endocrine disease affecting 29 million people, is characterized by insulin resistance that results in blood glucose (BG) elevations. Patients with long-term uncontrolled DM2 are at increased risk for multiple vascular complications, including nephropathy, neuropathy, vision and hearing loss, and heart disease. Ertugliflozin is the fourth SGLT2 inhibitor to be approved by the FDA for the treatment of DM2, the others being dapagliflozin (Farxiga), empagliflozin (Jardiance), and canagliflozin (Invokana). Ertugliflozin has also been approved in fixed-dose combinations with metformin (Segluromet) and sitagliptin (Steglujan).
The FDA’s approval of ertugliflozin was based on data from seven randomized, double-blind, placebo- and active comparator–controlled studies of 4,863 patients with DM2. The studies included ertugliflozin as monotherapy and add-on therapy, and all had a primary efficacy endpoint of reduction in glycosylated hemoglobin A1C (HbA1C) percentage on therapy. Results of the studies demonstrated statistically significant reductions in HbA1C in trials of ertugliflozin monotherapy (0.6%-0.7% reduction); as an add-on to metformin (0.5%-0.7% reduction); as an add-on to metformin plus sitagliptin (0.5%-0.6% reduction); and as triple therapy with metformin plus sitagliptin versus the individual components of the triple therapy (1%-1.4% reduction). It was also shown to be noninferior to glimepiride as an add-on to metformin therapy. Ertugliflozin did not demonstrate efficacy in patients with DM2 and comorbid moderate renal impairment.
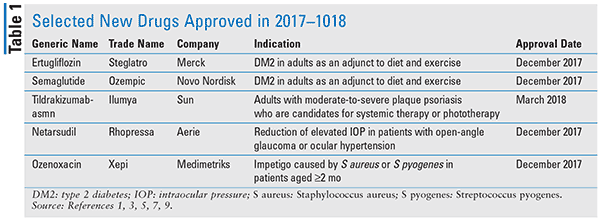
Pharmacology and Pharmacokinetics1,2: Ertugliflozin is a biaryl-sugar derivative (Figure 1) that inhibits SGLT2, a renal transport protein responsible for the majority of glucose reabsorption from the proximal renal tubule of the nephron. Inhibition of this transporter by ertugliflozin results in increased glucose excretion and an overall reduction in BG levels. The 15-mg formulation of ertugliflozin is approximately 100% orally bioavailable and has a mean half-life of 16.6 hours. The drug is 93.6% plasma protein–bound. It is primarily metabolized via O-glucuronidation and does not induce or inhibit CYP450 isoenzymes. About 41% of the dose is excreted in the feces and 50% in the urine.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions (5%) reported in clinical trials were female genital fungal infections. As an SGLT2 inhibitor, ertugliflozin carries the same warnings as others in the class, including the potential for elevations in LDL cholesterol, as well as an increased risk of ketoacidosis, acute kidney injury and renal failure, urosepsis and pyelonephritis, genital mycotic infections, lower-limb amputation, and severe hypotension. Ertugliflozin is not recommended in the second or third trimester of pregnancy because it may affect renal development during this time.
Ertugliflozin has no significant interactions with CYP450 substrates or inhibitors or with renally excreted drugs. Patients taking ertugliflozin in combination with insulin or an insulin secretagogue may have to lower the insulin or insulin secretagogue dosage to reduce the risk of hypoglycemia.
Dosage and Administration1,2: Ertugliflozin is supplied as 5 mg- and 15-mg tablets. The recommended dosage is 5 mg every morning, with or without food, which may be increased to 15 mg once daily in patients who require additional glycemic control. Ertugliflozin use is contraindicated in patients with an estimated glomerular filtration rate (eGFR) less than 30 mL/minute/1.73 m2, and it is not recommended in those with an eGFR less than 60 mL/minute/1.73 m2. No dosage adjustment is recommended in elderly patients or in patients with mild renal impairment or mild-to-moderate hepatic impairment. Ertugliflozin has not been studied in pediatric patients or in patients with severe hepatic impairment.
Semaglutide (Ozempic, Novo Nordisk)
Indication and Clinical Profile3,4: Semaglutide is a once-weekly, injectable glucagon-like peptide-1 (GLP-1) receptor agonist that has been approved for the treatment of type 2 diabetes (DM2) in adult patients as an adjunct to diet and exercise. DM2 is a metabolic disorder characterized by insulin resistance and reduced insulin secretion, which results in long-term high blood glucose (BG) levels. Over time, high BG levels can lead to a number of complications, including retinopathy, nephropathy, neuropathy, reduced circulation, and heart disease. Semaglutide is the sixth GLP-1 agonist to be approved by the FDA for the treatment of DM2; the others are exenatide (Byetta/Bydureon), liraglutide (Victoza), albiglutide (Tanzeum), dulaglutide (Trulicity), and lixisenatide (Adlyxin).
FDA approval of semaglutide was based on data from eight trials involving more than 8,000 subjects with DM2 that evaluated semaglutide 0.5 mg or 1 mg once weekly as monotherapy and in combination with metformin plus or minus a sulfonylurea, thiazolidinediones, and basal insulin. The primary endpoint of all trials was a reduction in glycosylated hemoglobin A1C (HbA1C) percentage on therapy. In placebo-controlled trials evaluating semaglutide used as monotherapy and as an add-on to insulin glargine, treatment with semaglutide resulted in statistically significant reductions in glycosylated HbA1C of –1.2% to –1.4% and –1.1% to –1.6% respectively, compared with placebo. In comparison studies versus sitagliptin 100 mg once daily, exenatide 2 mg once weekly, and titrated insulin glargine, semaglutide use resulted in statistically significant reductions in HbA1C (–0.6% to –0.8%; –0.5%; and 0.3% to –0.6% respectively) versus use of the comparative agents.
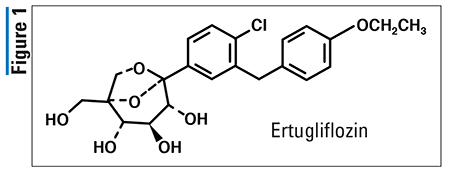
Pharmacology and Pharmacokinetics3,4: Semaglutide’s (Figure 2) therapeutic effect of reducing BG levels is achieved via its agonist activity at GLP-1 receptors present in the pancreas and stomach. Activation of pancreatic GLP-1 receptors suppresses glucagon secretion while stimulating glucose-dependent insulin secretion, resulting in a direct reduction of BG levels. The activation of GLP-1 receptors in the stomach causes delayed gastric emptying, which promotes satiety and reduces appetite. This drug is an analogue of GLP-1, where an aminoisobutyric acid group has been substituted at position 8 to protect the molecule from inactivation by dipeptidyl peptidase-4 (DPP-4), and a long-chain fatty acid has been connected to lysine 26 to enhance plasma protein binding and extend half-life.
Semaglutide achieves its peak plasma concentration 1 to 3 days after injection, with steady-state exposure achieved after 4 to 5 weeks of regular dosing. While protected from DPP-4 metabolism, semaglutide is metabolized slowly via beta-oxidation of its fatty-acid sidechain after proteolytic cleavage of the peptide backbone. It is predominantly excreted in urine and feces and has a mean terminal half-life of 7 days.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions reported by patients receiving semaglutide in clinical trials were nausea, vomiting, abdominal pain, constipation, and hypoglycemic events, the last of which was most common when used with basal insulin (16.7%-29.8%). The semaglutide drug label comes with a black box warning regarding its potential to cause thyroid C-cell tumors and highlights that semaglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in those with multiple endocrine neoplasia syndrome type 2. Use of semaglutide may result in acute kidney injury, worsening of chronic renal failure, severe hypersensitivity reactions, and pancreatitis. Patients at high cardiovascular risk who are receiving semaglutide may be at increased risk for complications from diabetic retinopathy and should be monitored.
Results of animal reproductive studies have shown that there may be a potential risk of early pregnancy loss and smaller birthweight from exposure to semaglutide during pregnancy. Therefore, it should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Patients using semaglutide in combination with sulfonylureas and insulins are at increased risk for hypoglycemia and may need to have their dose reduced when starting treatment with semaglutide. Because of semaglutide’s ability to delay gastric emptying, caution should be exercised when oral medications are concomitantly administered with semaglutide, as it may impact the absorption of those medications.
Dosage and Administration3,4: Semaglutide is supplied as a prefilled pen for SC injection at a concentration of 2 mg/1.5 mL. It is available as two different pens configured to deliver either 1 mg or 0.25 mg and 0.5 mg per injection. Recommended dosing is 0.25 mg SC once weekly for 4 weeks followed by 0.5 mg SC once weekly, up to a maximum dosage of 1 mg once weekly. Safety and efficacy of semaglutide have not been studied in pediatric patients. Because of potential fetal risk and a long washout period, women receiving semaglutide should discontinue the medication at least 2 months prior to a planned pregnancy. Semaglutide does not require dose adjustments for patients with renal or hepatic impairment.
Tildrakizumab-asmn (Ilumya, Sun)
Indication and Clinical Profile5,6: Tildrakizumab-asmn is specifically indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Plaque psoriasis, a chronic autoimmune disorder characterized by the generation of thick, scaly patches of skin, is the most common form of psoriasis. The skin plaques appear as red, raised areas covered with flaky white scales, and they are often itchy and painful.
FDA approval of tildrakizumab-asmn was based on data from two multicenter, randomized, double-blind, placebo-controlled trials including 926 subjects with moderate-to-severe plaque psoriasis with a Physician’s Global Assessment (PGA) score of 3. The primary endpoint of both trials was the achievement of at least 75% of skin clearance, as measured by the Psoriasis Area Sensitivity Index (PASI) and a PGA score of 0 or 1 at Week 12 after two doses. At Week 12, 61% to 64% of patients treated with tildrakizumab-asmn 100 mg achieved a PASI 75 and 55% to 58% achieved a PGA score of 0 or 1, a significant improvement over those treated with placebo (6% achieved PASI 75 and 4% to 7% achieved PGA 0 or 1).
Pharmacology and Pharmacokinetics5,6: Tildrakizumab-asmn is a humanized immunoglobulin G1 kappa monoclonal antibody that acts by binding to interleukin 23 (IL-23), a proinflammatory cytokine involved in immune response. By selectively binding to IL-23, tildrakizumab-asmn prevents IL-23 from interacting with its receptor, thereby inhibiting the release of proinflammatory cytokines and chemokines mediated by IL-23.
Tildrakizumab-asmn has an absolute bioavailability of 73% to 80% following injection and achieves its peak plasma concentration 6 days after injection. Tildrakizumab-asmn’s metabolism is expected to be degradation to small peptides via catabolic pathways in a manner similar to endogenous immunoglobulin G. It is cleared at a mean rate of 0.32 L/day and has a half-life of approximately 23 days.
Adverse Reactions and Drug Interactions5,6: The most common (1%) adverse reactions reported by patients receiving tildrakizumab-asmn in clinical trials were upper respiratory infections, injection-site reactions, and diarrhea. Other adverse reactions noted during clinical trials included dizziness and pain in extremities, as well as rare cases of angioedema and urticaria. Tildrakizumab-asmn may increase the risk of infection and should not be initiated in patients with an untreated, active infection or in patients who have not had tuberculosis infection ruled out through evaluation. Patients treated with tildrakizumab-asmn who develop a clinically important infection or who are not responding to standard therapy should be closely monitored and should discontinue tildrakizumab-asmn until the infection resolves. The formation of antibodies to tildrakizumab-asmn, including neutralizing antibodies, has been experienced in 6.5% of patients and may result in reduced serum concentrations of tildrakizumab-asmn and reduced efficacy.
Because of its potential to increase risk of infection, avoid use of live vaccines in patients treated with tildrakizumab-asmn. Patients should receive all age-appropriate immunizations according to current immunization guidelines prior to initiating therapy.
Dosage and Administration5,6: Tildrakizumab-asmn is supplied as a single-use, prefilled syringe containing 1 mL of a 100-mg/mL solution. The recommended dosage is 100 mg at weeks 0 and 4, and every 12 weeks thereafter. The safety and efficacy of tildrakizumab-asmn has not been studied in pediatric patients or patients with renal or hepatic impairment. It is contraindicated in patients with a previous serious hypersensitivity reaction to tildrakizumab-asmn or to any of the components of the drug.
Netarsudil (Rhopressa, Aerie)
Indication and Clinical Profile7,8: Netarsudil ophthalmic solution is specifically indicated for the reduction of elevated intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension. It is the first new drug class in more than two decades introduced for the reduction of IOP.
The efficacy of netarsudil was assessed in three completed phase III clinical trials (Rocket 1, 2, and 4) comparing either once daily or twice daily netarsudil administration with timolol administered twice daily. In Rocket 2, a 12-month safety trial with a 90-day efficacy assessment, netarsudil dosed both once daily and twice daily achieved its primary efficacy endpoint demonstrating noninferiority compared with twice-daily timolol. The primary efficacy endpoint evaluated subjects with prestudy baseline IOPs of above 20 to below 25 mmHg. Data demonstrated a consistent level of IOP lowering across all baseline IOPs and throughout the 90-day efficacy period. Similar results were achieved in the Rocket 4 trial (duration of 6 months) comparing once-daily netarsudil to twice-daily timolol. Rocket 1 was a 90-day efficacy trial that did not achieve its primary endpoint, but netarsudil did demonstrate noninferiority compared with timolol in subjects with IOP below 26 mmHg at all nine measured time points and numerical superiority over timolol at the majority of measured time points. Rocket 3 is a 12-month safety-only study in Canada that is currently in progress.
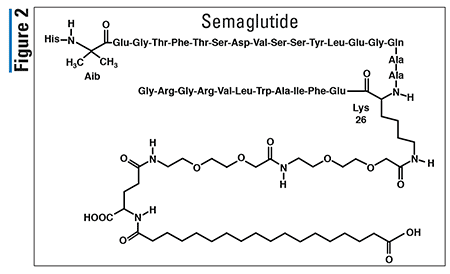
Pharmacology and Pharmacokinetics7,8: Netarsudil is an isoquinolone-amide derivative that functions as a Rho kinase inhibitor (Figure 3). Inhibition of this kinase is believed to reduce IOP by increasing the outflow of aqueous humor through the trabecular meshwork route, the main fluid drain of the eye. The exact mechanism is unknown.
After topical ocular dosing, netarsudil is metabolized by esterases in the eye to an active metabolite designated as AR-13503 (Figure 3). In clinical trials, no quantifiable plasma concentrations of netarsudil or its active metabolite were detected, suggesting very minimal system exposure. Therefore, data regarding drug distribution, metabolism, and excretion are not available.
Adverse Reactions and Drug Interactions7,8: The most common adverse event reported in clinical trials was hyperemia, which occurred in 53% of patients. Other common (approximately 20%) ocular adverse reactions reported were corneal verticillata, instillation-site pain, and conjunctival hemorrhage. Instillation- site erythema, corneal staining, blurred vision, increased lacrimation, erythema of the eyelid, and reduced visual acuity were reported in 5% to 10% of patients. It should be noted that there have been reports of bacterial keratitis associated with the use of multiple-dose containers of topical ophthalmic products. These containers had been inadvertently contaminated by patients who, in most cases, had a concurrent corneal disease or a disruption of the ocular epithelial surface. There are no available data on netarsudil use in pregnancy or during breastfeeding to inform any drug-associated risk of adverse developmental outcomes. However, systemic exposure to netarsudil from ocular administration is low.
Ozenoxacin (Xepi, Medimetriks)
Indication and Clinical Profile9,10: Ozenoxacin is a new quinolone antibiotic cream indicated for the treatment of impetigo caused by Staphylococcus aureus or Streptococcus pyogenes in patients aged 2 months and older. Impetigo is a contagious bacterial skin infection that most commonly affects infants, young children, and those involved in close-contact sports or living in enclosed environments. Impetigo is estimated to account for approximately 10% of the skin problems observed in pediatric clinics in the United States and is considered the most common bacterial skin infection and a significant public-health concern. Ozenoxacin is the first new topical treatment for impetigo in more than 10 years in a market that generates more than 8 million prescriptions annually.
The approval of ozenoxacin is based on a clinical-development program that includes the results of two phase III, multicenter, controlled trials that enrolled 723 subjects aged 2 months and older with impetigo. Subjects with underlying skin disease, skin trauma, clinical evidence of secondary infection, or systemic signs and symptoms of infection (such as fever) were excluded from these studies. Ozenoxacin demonstrated superiority versus placebo on the prespecified clinical and bacteriological endpoints when applied topically twice daily for 5 days. Bacterial success, defined as bacterial eradication or presumed eradication, was achieved at the end of treatment in 90.8% of patients using ozenoxacin versus 69.8% of those given placebo (P <.0001). Ozenoxacin showed excellent antibacterial activity against S pyogenes and S aureus, including methicillin-resistant S aureus.
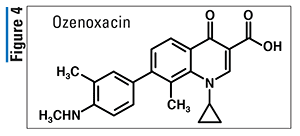
Pharmacology and Pharmacokinetics9,10: Ozenoxacin belongs to a new generation of nonfluorinated, nonpiperazine quinolones (Figure 4). Its antibacterial mechanism of action involves the inhibition of bacterial DNA-replication enzymes, DNA gyrase A and topoisomerase IV. Ozenoxacin has been shown to be bactericidal against S aureus and S pyogenes organisms. Ozenoxacin is not significantly absorbed from administration sites; therefore, data regarding drug distribution, metabolism, and excretion are not available.
Adverse Reactions and Drug Interactions9,10: In clinical trials, ozenoxacin was found to be negligibly absorbed, safe, and well tolerated in pediatric and adult patients aged 2 months and older. The primary adverse effects were rosacea and seborrheic dermatitis. Prolonged use of ozenoxacin may result in overgrowth of nonsusceptible bacteria and fungi. If such infections occur, discontinue use and institute alternative therapy. The safety profile of ozenoxacin in pediatric patients aged 2 months and older was similar to that in adults. The safety and effectiveness of ozenoxacin in pediatric patients aged younger than 2 months have not been established. There are no available data on the use of ozenoxacin in pregnancy or breastfeeding to inform a drug-associated risk of adverse developmental outcomes.
Dosage and Administration9,10: Ozenoxacin is supplied as a 1% pale-yellow cream for topical administration. Each gram contains 10 mg of ozenoxacin. A thin layer of ozenoxacin is applied topically to the affected area twice daily for 5 days. The affected area may be up to 100 cm2 in adult and pediatric patients aged 12 years and older, or 2% of the total body surface area and not exceeding 100 cm2 in pediatric patients aged less than age 12 years. Ozenoxacin is not for oral, ophthalmic, intranasal, or intravaginal use.
REFERENCES
1. Steglatro (ertugliflozin) package insert. Whitehouse Station, NJ: Merck and Co, Inc; December 2017.
2. Aronson R, Frias J, Goldman A, et al. Long-term efficacy and safety of ertugliflozin monotherapy in patients with inadequately controlled T2DM despite diet and exercise: VERTIS MONO extension study. Diabetes Obes Metab. 2018;20(6):1453-1460.
3. Ozempic (semaglutide) package insert. Plainsboro, NJ: Novo Nordisk Inc; December 2017.
4. Davies M, Pieber TR, Hartoft-Nielsen ML, et al. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes. JAMA. 2017;318(15):1460-1470.
5. Ilumya (tildrakizumab-asmn) package insert. Cranbury, NJ: Sun Pharmaceuticals Inc; March 2018.
6. Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017:390(10091):276-288.
7. Rhopressa (netarsudil) package insert. Irvine, CA: Aerie Pharmaceuticals, Inc; December 2017.
8. Aerie Pharmaceuticals. Aerie pharmaceuticals initiates fourth phase 3 clinical trial of Rhopressa. September 24, 2015. http://investors.aeriepharma.com/releasedetail.cfm?ReleaseID=932929. Accessed June 24, 2018.
9. Xepi (ozenoxacin) package insert. Fairfield, NJ: Medimetriks Pharmaceuticals Inc; December 2017.
10. López Y, Tato M, Espinal P, et al. in vitro activity of ozenoxacin against quinolone-susceptible and quinolone-resistant gram-positive bacteria. Antimicrob Agents Chemother. 2013;57(12):6389-6392.
To comment on this article, contact rdavidson@uspharmacist.com.



