US Pharm. 2021;46(10):HS1-HS10.
New molecular entities (NMEs) are defined by the FDA as new drug products containing a chemical substance marketed for the first time in the United States as their active ingredient. The following descriptions of NMEs approved in 2020-2021 (TABLE 1) detail the basic pharmacologic and clinical profile for each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse reaction, drug interaction and dosing data submitted to the FDA in support of the manufacturer’s new drug application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile, not detected in premarketing studies, emerge after the drug is used in the population in general. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years of their first becoming available. Some of these drugs may eventually acquire black box warnings for serious adverse drug reactions or be withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers an initial assessment of newly approved drugs, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by patients.
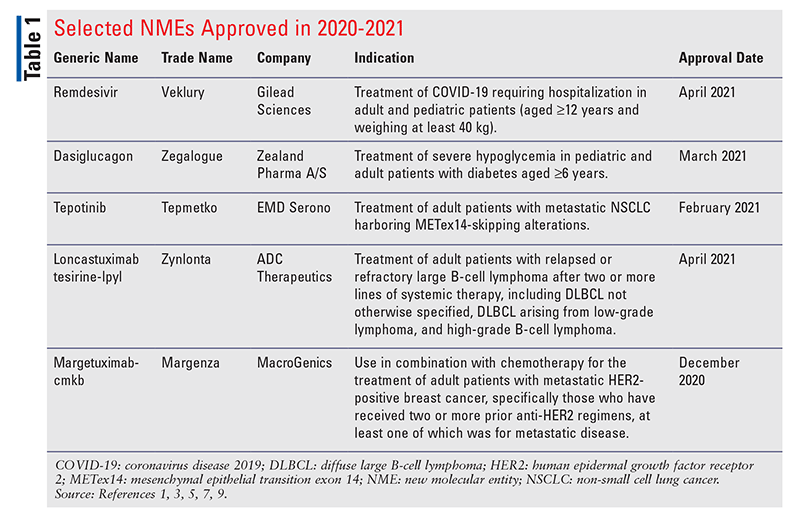
Remdesivir (Veklury, Gilead Sciences)
Indication and Clinical Profile1,2: Remdesivir is indicated for the treatment of coronavirus disease 2019 (COVID-19) requiring hospitalization in adult and pediatric patients (at least 12 years of age and weighing at least 40 kg). The FDA approval of remdesivir was based on several clinical trials. One of these, the Adaptive Covid-19 Treatment Trial (ACTT-1), was a global, randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of a 10-day treatment course of remdesivir versus placebo in 1,063 hospitalized adult patients. Trial patients had confirmed SARS-CoV-2 infection with mild, moderate, or severe symptoms and were also receiving standard-of-care treatment. The primary outcome measure was time to recovery within 29 days after randomization. Recovery was defined as discharge from the hospital without limitations on activities, discharge from the hospital with limitations on activities and/or requiring home oxygen, or hospitalized but not requiring supplemental oxygen or ongoing medical care. Remdesivir significantly improved time to recovery as compared with placebo by 5 days in the study population (10 vs. 15 days) and 7 days in patients who required oxygen support at baseline (11 vs. 18 days). As a secondary endpoint, remdesivir also reduced disease progression in patients needing oxygen, resulting in a significantly lower incidence of new mechanical ventilation or extracorporeal membrane oxygenation (13% vs. 23%). In the overall patient population, there was reduced mortality with remdesivir compared with placebo at Day 29 (11.4% vs. 15.2%).
Pharmacology and Pharmacokinetics1,2: Remdesivir is a phosphoramide nucleoside prodrug (FIGURE 1) that functions as an inhibitor of the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp), which is essential for viral replication. The drug distributes into infected cells where it is metabolized to the corresponding nucleoside remdesivir monophosphate intermediate by carboxyesterase 1 and/or cathepsin A. The monophosphate is subsequently phosphorylated by cellular kinases to form the pharmacologically active triphosphate metabolite. Remdesivir triphosphate (RDV-TP) is an analog of adenosine triphosphate (ATP) and competes with high selectivity (3.65-fold) over the natural ATP substrate for incorporation into growing viral RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase. This action results in chain termination during replication of the viral RNA. In a biochemical assay, RDV-TP inhibited RNA synthesis with an IC50 value of 0.032 µM. RDV-TP also inhibits viral RNA synthesis following its incorporation into the template viral RNA as a result of read-through by the viral polymerase that may occur at higher nucleotide concentrations. When remdesivir nucleotide is present in the viral RNA template, the efficiency of incorporation of the complementary natural nucleotide is compromised, thereby inhibiting viral RNA synthesis. RDV-TP specificity results from its relatively weak inhibition of mammalian DNA and RNA polymerases, including human mitochondrial RNA polymerase.
RDV exhibits a linear pharmacokinetic profile following single-dose IV administration. High intracellular concentrations of the active triphosphate RDV-TP (220- to 370-fold higher than the in vitro half-maximal effective concentration against SARS-CoV-2 clinical isolate) were achieved following infusion of 75-mg or 150-mg doses of the drug. Following multiple doses of RDV 150 mg once daily for 7 to 14 days, RDV exhibited a pharmacokinetic profile similar to single-dose administration.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions associated with the use of remdesivir include nausea and increases in transaminases (alanine aminotransferase [ALT] and aspartate transaminase [AST]). Transaminase elevations have been observed in patients with COVID-19 who received remdesivir and as a clinical feature of COVID-19. Thus, hepatic testing should be performed in all patients receiving this drug. Also, it may be necessary to discontinue remdesivir if ALT levels increase to >10x ULN, and if ALT elevation is accompanied by signs or symptoms of liver inflammation. Hypersensitivity, including infusion-related and anaphylactic reactions, has been observed. Slower infusion rates, with a maximum infusion time of up to 120 minutes, may prevent signs and symptoms of hypersensitivity. If symptoms of a clinically significant hypersensitivity reaction occur, remdesivir administration should be discontinued promptly and appropriate treatment initiated.
Drug-drug interaction studies with remdesivir have not been performed in humans. While the drug and its metabolites are in vitro substrates and/or inhibitors of certain drug-metabolizing enzymes and transporters, the clinical relevance of these in vitro assessments has not been established. Coadministration of remdesivir with chloroquine phosphate or hydroxychloroquine sulfate is not recommended due to antagonism observed in cell culture, which may lead to a decrease in antiviral activity of remdesivir.
Dosage and Administration1,2: Remdesivir is supplied as an injection for IV administration, and dilution is required prior to infusion. This drug should only be administered in a healthcare setting capable of providing acute care. The recommended dosage for adults and pediatric patients aged 12 years and older and weighing at least 40 kg is a single loading dose of 200 mg on Day 1, followed by once-daily maintenance doses of 100 mg from Day 2. The recommended treatment duration for patients not requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO) is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days (total treatment maximum duration of 10 days). Laboratory tests including eGFR, hepatic enzymes, and prothrombin time should be performed prior to initiating remdesivir and during use as appropriate. Remdesivir is not recommended in individuals with eGFR less than 30 mL/min.
Dasiglucagon (Zegalogue, Zealand Pharma A/S)
Indication and Clinical Profile3,4: Dasiglucagon is indicated for the treatment of severe hypoglycemia in pediatric and adult patients with diabetes aged 6 years and older. Three randomized, double-blind, placebo-controlled, multicenter trials were conducted in patients with type 1 diabetes; two trials (Trial A and Trial B) were conducted in adult patients, and one trial (Trial C) was conducted in pediatric patients aged 6 to 17 years. In all three trials, patients were randomized to dasiglucagon 0.6 mg, placebo, or glucagon for injection 1.0 mg. Dasiglucagon and the comparators were administered as single SC injections following a controlled induction of hypoglycemia with IV insulin. During this procedure, a plasma glucose concentration less than 60 mg/dL was targeted in Trials A and B, whereas the target was less than 80 mg/dL in Trial C. The primary efficacy endpoint for all three trials was time to plasma glucose recovery, defined as an increase in blood glucose of 20 mg/dL or greater from time of administration, without additional intervention within 45 minutes. In Trial A the median time to plasma glucose recovery was statistically significantly shorter for dasiglucagon (10 minutes) versus placebo (40 minutes). The median time to plasma glucose recovery was numerically similar between dasiglucagon (10 minutes) and glucagon for injection (12 minutes). Similar results were obtained in the other trials. It should be noted that dasiglucagon is effective in treating hypoglycemia only if sufficient hepatic glycogen is present. Patients in states of starvation, with adrenal insufficiency, or with chronic hypoglycemia may not have adequate levels of hepatic glycogen for this drug to be effective. Patients with these conditions should be treated with glucose.
Pharmacology and Pharmacokinetics3,4: Dasiglucagon contains dasiglucagon hydrochloride, which is a 29 amino acid glucagon analog and glucagon receptor agonist. This drug increases blood-glucose concentration by activating hepatic glucagon receptors, thereby stimulating glycogen breakdown and release of glucose from the liver. Thus, hepatic stores of glycogen are necessary for dasiglucagon to produce an antihypoglycemic effect.
Subcutaneous injection of 0.6 mg dasiglucagon results in a mean peak plasma concentration of 5110 pg/mL at around 35 minutes. The mean apparent volume of distribution is 47-57 L. Metabolism studies suggest that dasiglucagon is cleared by proteolytic degradation pathways in blood and multiple organs, similar to glucagon. The half-life is approximately 30 minutes.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (greater than or equal to 2%) reported with dasiglucagon in adults include nausea, vomiting, headache, diarrhea, and injection-site pain. In pediatric patients, nausea, vomiting, headache, and injection-site pain were observed most often. Hypersensitivity reactions have been reported with glucagon products and may include generalized rash, but in some cases, anaphylactic shock with breathing difficulties and hypotension can occur. Dasiglucagon is contraindicated in patients with pheochromocytoma because it may stimulate the release of catecholamines from the tumor and significantly increase the risk for severe hypertension. It is also contraindicated in patients with insulinoma because it may stimulate exaggerated insulin release from an insulinoma and cause subsequent hypoglycemia.
In animal reproduction studies, daily SC administration of dasiglucagon did not cause adverse developmental effects even at exposures significantly higher than the human dose. There are no available data on dasiglucagon use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. However, untreated hypoglycemia in pregnancy can cause complications and may be fatal. There is no information on the presence of dasiglucagon in either human or animal milk, or the effects of the drug on the breastfed infant or milk production. Dasiglucagon is a peptide and would be expected to be metabolically inactivated in the infant’s digestive tract and therefore unlikely to cause harm.
Limited data are currently available concerning drug interactions with dasiglucagon. The drug may increase the anticoagulant effect of warfarin. Patients taking beta-blockers may have a transient increase in pulse and blood pressure. In patients taking indomethacin, dasiglucagon may lose its ability to raise blood glucose or may produce hypoglycemia.
Dosage and Administration3,4: Dasiglucagon is supplied as a 0.6-mg/0.6-mL single-dose autoinjector and prefilled syringes for SC injection only. The recommended dose in adults and pediatric patients aged 6 years and older is 0.6 mg administered into the lower abdomen, buttocks, thigh, or outer upper arm. If there is no response after 15 minutes, an additional 0.6-mg dose from a new device may be administered while waiting for emergency assistance.
Tepotinib (Tepmetko, EMD Serono)
Indication and Clinical Profile5,6: Tepotinib is a kinase inhibitor indicated for the treatment of adult patients with metastatic non–small cell lung cancer (NSCLC) harboring mesenchymal epithelial transition exon 14 (METex14) skipping alterations. Tepotinib received accelerated approval based on overall response rate (ORR) and duration of response (DOR) in trials to date. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial.
The approval of tepotinib was based on results from the pivotal phase II VISION study, an ongoing multicenter, nonrandomized, open-label study investigating the drug as monotherapy in 152 patients with advanced or metastatic NSCLC with METex14-skipping alterations. Patients also were required to have epidermal growth factor receptor (EGFR) wild-type and anaplastic lymphoma kinase (ALK)-negative status, at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1. Patients received tepotinib 450 mg once daily until disease progression or unacceptable toxicity. In this study, tepotinib demonstrated an ORR of 43% in both treatment-naïve and previously treated patients (n = 83). Median DOR was 10.8 months and 11.1 months among treatment-naïve and previously treated patients, respectively. DOR of 6 months or more occurred among 67% of treatment-naïve patients and 75% of previously treated patients, and DOR of 9 months or more occurred among 30% of treatment-naïve patients and 50% of previously treated patients.
Pharmacology and Pharmacokinetics5,6: Tepotinib is a dihydropyridazin-3-benzonitrile MET kinase inhibitor (FIGURE 2) that targets variants with exon 14–skipping alterations. This drug inhibits hepatocyte growth factor (HGF)-dependent and independent MET phosphorylation and MET-dependent downstream signaling pathways. Tepotinib also inhibits melatonin 2 and imidazoline 1 receptors at clinical concentrations. In vitro, tepotinib inhibits tumor cell proliferation, anchorage-independent growth, and migration of MET-dependent tumor cells.
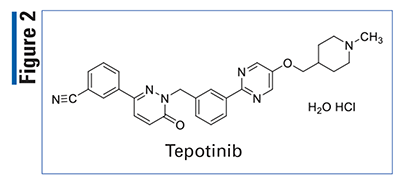
The bioavailability of tepotinib in the fed state is approximately 70%. It has an apparent volume of distribution of 1,038 L and is 98% bound by plasma proteins. Tepotinib is metabolized primarily by CYP3A4 and CYP2C8 and excreted as the parent drug and metabolites primarily in the feces (85%). The apparent clearance is 23.8 L/hour with a half-life of 32 hours.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions (greater than or equal to 20%) associated with tepotinib include edema, fatigue, nausea, diarrhea, musculoskeletal pain, and dyspnea. The most common Grade 3 to 4 laboratory abnormalities (greater than or equal to 2%) were decreased hemoglobin, lymphocytes, albumin, sodium, and increased gamma-glutamyltransferase, amylase, ALT and AST. Interstitial lung disease (ILD)/pneumonitis and hepatotoxicity have been reported in patients treated with tepotinib. If patients develop new or worsening pulmonary symptoms indicative of ILD/pneumonitis or hepatotoxicity, the drug should be withheld and may need to be discontinued per manufacturer’s guidelines. Based on observations in animal studies and the drug’s mechanism of action, tepotinib can cause fetal harm when administered to pregnant women. Thus females of reproductive potential or males with female partners of reproductive potential should be advised to use effective contraception during treatment and for one week after the final dose. Also, women should not breastfeed during treatment and for one week after the final dose.
Metabolic studies suggest concomitant use of drugs that are strong CYP3A inhibitors and P glycoprotein (P-gp) inhibitors may increase tepotinib exposure resulting in an increase in the incidence and severity of adverse reactions. Thus, it should not be used with dual strong CYP3A and P-gp inhibitors. Tepotinib should not be used concurrently with strong CYP3A inducers since this may lead to drug inactivation and loss of efficacy. Also, since tepotinib is a P-gp inhibitor, it should be avoided in patients receiving concurrent therapy with P-gp substrates where minimal concentration changes may lead to serious or life-threatening toxicities.
Dosage and Administration5,6: Tepotinib is supplied as 225-mg tablets for oral administration. The recommended dosage is 450 mg orally once daily with food until disease progression or unacceptable toxicity. The drug should be taken at approximately the same time every day and tablets swallowed whole; tablets should not be chewed, crushed, or split. If vomiting occurs after taking a dose, patients are advised to take the next dose at the scheduled time. Missed doses within 8 hours of the next scheduled dose should not be made up.
No dosage modification is recommended in patients with mild or moderate renal impairment or hepatic impairment. The recommended dosage has not been established for patients with severe renal impairment, and safety of tepotinib in patients with severe hepatic impairment has not been investigated.
Loncastuximab Tesirine-lpyl (Zynlonta, ADC Therapeutics)
Indication and Clinical Profile7,8: Loncastuximab tesirine-lpyl is an antibody-alkylating agent conjugate that is specifically indicated for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. DLBCL is an aggressive type of non-Hodgkin lymphoma (NHL) that affects B-lymphocytes. It is the most common type of NHL in the world, accounting for about 22% of newly diagnosed cases of B-cell NHL in the United States. More than 18,000 people are diagnosed with DLBCL each year. FDA approval of loncastuximab tesirine-lpyl was based on data from LOTIS-2, a phase II multinational, single-arm clinical trial involving 145 adult patients with relapsed or refractory DLBCL following two or more prior lines of systemic therapy. In this trial loncastuximab tesirine-lpyl treatment resulted in an overall response rate (ORR) of 48.3% (70/145 patients), which included a complete response rate of 24.1% and a partial response rate of 24.1%. Patients had a median time to response of 1.3 months, and the median duration of response for the 70 responders was 10.3 months (inclusive of patients who were censored). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial or trials.
Pharmacology and Pharmacokinetics7,8: Loncastuximab tesirine-lpyl is an antibody-drug conjugate (ADC) targeting CD19, a transmembrane protein expressed on the surface of cells of B-lineage. The monoclonal IgG1 kappa antibody component binds to human CD19. The small molecule component (SG3199) is a PBD dimer and alkylating agent. Upon binding to CD19, the drug is internalized, followed by release of SG3199 via proteolytic cleavage. The released SG3199 binds to the DNA minor groove and forms highly cytotoxic DNA interstrand crosslinks, inducing cell death.
After IV administration the mean volume of distribution of loncastuximab tesirine-lpyl is 7.11 L. The monoclonal antibody portion (loncastuximab) is expected to be metabolized into small peptides by normal protein catabolism. The cytotoxin, SG3199, is metabolized by CYP3A4/5. The major excretion pathways of SG3199 have not been studied in humans, but it is expected to be minimally renally excreted. The mean clearance of loncastuximab tesirine-lpyl decreases with time from 0.499 L/day (89.3%) after a single dose to 0.275 L/day (38.2%) at steady state. The mean half-life is approximately 20 days at steady state.
Adverse Reactions and Drug Interactions7,8: The most common (≥20%) adverse reactions associated with loncastuximab tesirine-lpyl treatment include thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain. Patients receiving this medication should be monitored for the development of pleural effusion, pericardial effusion, ascites, peripheral edema, myelosuppression, cutaneous reactions, and infections. It may be necessary to withhold, reduce, or discontinue the medication if these conditions emerge. Loncastuximab tesirine-lpyl contains a genotoxic compound (SG3199) that can cause embryo-fetal harm. Patients should be advised of the potential risk to a fetus and to use effective contraception. There are no data on the presence of this antibody or SG3199 in human milk, the effects on the breastfed child, or milk production.
The cytotoxic component of loncastuximab tesirine-lpyl, SG3199, does not inhibit any major cytochrome isoform, or transporters including P-glycoprotein (P-gp), BCRP, OATP1B1, OATP1B3, organic anion transporters (OAT1 and 3, OCT2, OCT1), multi-antimicrobial extrusion protein (MATE1 and 2-K) at clinically relevant concentrations. SG3199 is a substrate of P-gp, but not a substrate of breast cancer resistance protein (BCRP), OATP1B1, or OCT1. No known drug interactions have been reported to date.
Dosage and Administration7,8: Loncastuximab tesirine-lpyl is supplied as 10 mg of a lyophilized powder in a single-dose vial for reconstitution and further dilution prior to IV administration. The recommended infusion dose is 0.15 mg/kg every 3 weeks for 2 cycles and 0.075 mg/kg every 3 weeks for subsequent cycles. Each infusion is administered over 30 minutes on Day 1 of each cycle. Patients should be premedicated with dexamethasone (4 mg orally or IV) twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl administration. This drug has not been studied in patients with moderate or severe hepatic impairment, but no dose adjustment is recommended for patients with mild hepatic impairment.
Margetuximab-cmkb (Margenza, MacroGenics)
Indication and Clinical Profile9,10: Margetuximab-cmkb is a monoclonal antibody indicated for use in combination with chemotherapy for the treatment of adult patients with metastatic human epidermal growth factor receptor 2 (HER2)-positive breast cancer, specifically those who have received two or more prior anti-HER2 regimens, at least one of which was for metastatic disease. FDA approval was based on SOPHIA, a randomized, open-label phase III clinical trial evaluating margetuximab-cmkb plus chemotherapy compared with trastuzumab plus chemotherapy in patients with HER2-positive metastatic breast cancer, who have previously been treated with anti-HER2-targeted therapies (trastuzumab, pertuzumab, and ado-trastuzumab emtansine or T-DM1). The study enrolled 536 patients who were randomized 1:1 to receive either margetuximab-cmkb given IV at 15 mg/kg every 3 weeks or trastuzumab given IV at 6 mg/kg (or 8 mg/kg for loading dose) every 3 weeks in combination with one of four chemotherapy agents (capecitabine, eribulin, gemcitabine, or vinorelbine) given at the standard doses. Intent-to-treat progression free survival (PFS) analysis occurred after 265 PFS events. The study showed a statistically significant 24% reduction in the risk of disease progression or death with margetuximab-cmkb plus chemotherapy compared with trastuzumab plus chemotherapy (median PFS 5.8 vs. 4.9 months). The objective response rate for margetuximab-cmkb plus chemotherapy was 22% and for trastuzumab plus chemotherapy was 16%.
Pharmacology and Pharmacokinetics9,10: Margetuximab-cmkb is an Fc-engineered, monoclonal antibody that targets the HER2 oncoprotein expressed by tumor cells in breast, gastroesophageal, and other solid tumors. The antibody inhibits tumor cell proliferation, reduces shedding of the HER2 extracellular domain and mediates antibody-dependent cellular cytotoxicity (ADCC), a mechanism similar to that of trastuzumab. In vitro, the modified Fc region of margetuximab-cmkb increases binding to the activating Fc receptor FCGR3A (CD16A) and decreases binding to the inhibitory Fc receptor FCGR2B (CD32B). These changes lead to greater in vitro ADCC and NK cell activation.
At the recommended dosage, the steady-state mean Cmax of margetuximab-cmkb is 466 µg/mL and AUC0-21d is 4120 µg.day/mL. The drug undergoes both linear and nonlinear elimination, and the time to steady state is estimated to be 2 months. Margetuximab-cmkb is presumed to be metabolized to small peptides by catabolic pathways, with a terminal half-life of 19.2 days and a clearance of 0.22 L/day (24%).
Adverse Reactions and Drug Interactions9,10: The most common adverse drug reactions (>10%) with margetuximab-cmkb in combination with chemotherapy are fatigue/asthenia, nausea, diarrhea, vomiting, constipation, headache, pyrexia, alopecia, abdominal pain, peripheral neuropathy, arthralgia/myalgia, cough, decreased appetite, dyspnea, infusion-related reactions, palmar-plantar erythrodysesthesia, and extremity pain. The margetuximab-cmkb label contains a black box warning concerning reductions in left ventricular ejection fraction associated with the drug. Thus, monitoring cardiac function prior to and during treatment is necessary, and treatment should be discontinued when a clinically significant decrease in left ventricular function occurs. Patients who receive anthracyclines less than 4 months after stopping margetuximab-cmkb may be at increased risk of cardiac dysfunction. Also, there is a warning regarding margetuximab-cmkb use during pregnancy since the drug can cause embryo-fetal harm. Patients should be advised of the risk and need for effective contraception.
Dosage and Administration9,10: Margetuximab-cmkb is supplied as a 250-mg/10-mL (25-mg/mL) solution for IV administration. The recommended dose is 15 mg/kg, administered as an IV infusion every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. The infusion should be administered over 120 minutes for the initial dose, then over a minimum of 30 minutes every 3 weeks for all subsequent doses. On days when both margetuximab-cmkb and chemotherapy are to be administered, margetuximab-cmkb may be administered immediately after completion of chemotherapy. Refer to the respective prescribing information for each therapeutic agent administered in combination with margetuximab-cmkb for the recommended dosage information, as appropriate.
REFERENCES
1. Veklury (remdesivir) package insert. Foster City, CA: Gilead Sciences, Inc; April 2021.
2. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19—final report. N Engl J Med. 2020;383(19):1813-1826.
3. Zegalogue (dasiglucagon) package insert. Søborg, Denmark: Zealand Pharma A/S; March 2021.
4. Hövelmann U, Olsen MB , Mouritzen U, et al. Low doses of dasiglucagon consistently increase plasma glucose levels from hypoglycaemia and euglycaemia in people with type 1 diabetes mellitus. Diabetes Obes Metab. 2019;21(3):601-610.
5. Tepmetko (tepotinib) package insert. Rockland, MA: EMD Serono, Inc; February 2021.
6. Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383(10):931-943.
7. Zynlonta (loncastuximab tesirine-lpyl) prescribing information. Murray Hill, NJ: ADC Therapeutics America; April 2021.
8. Caimi PF, Ai W, Alderuccio JP, et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021;22(62):790-800.
9. Margenza (margetuximab-cmkb) prescribing information. Rockville, MD: MacroGenics, Inc. December 2020.
10. Rugo HS, Im SA, Cardoso F, et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2021;7(4):573-584.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



