US Pharm. 2020;45(10):26-33.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2019–2020 (TABLE 1) details the basic clinical and pharmacologic profile of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Trifarotene (Aklief, Galderma)
Indication and Clinical Profile1,2: Trifarotene is the first new retinoid in more than 20 years to receive FDA approval for treating acne. It is indicated for the topical treatment of acne vulgaris in patients aged 9 years and older and is the first topical therapy specifically proven to treat facial and truncal acne. Acne is the most common skin disease in the United States, annually affecting up to 50 million Americans and approximately 85% of young people. This inflammatory disease occurs when sebum and dead skin cells clog pores, enabling the proliferation of Cutibacterium acnes. Facial acne is the most common and often most visible presentation, but more than one-half of people with facial acne (52%) also have truncal acne. Acne can trigger feelings of depression, poor body image, and low self-esteem.
FDA approval was based on data from two pivotal phase III clinical trials of once-daily trifarotene cream in patients with moderate acne on the face and trunk. The two identical 12-week, randomized, multicenter, vehicle-controlled clinical trials enrolled 2,420 patients. Compared with vehicle, trifarotene was found to significantly reduce inflammatory lesions on the face in as soon as 2 weeks and on the back, shoulders, and chest in 4 weeks (P <.05).
Pharmacology and Pharmacokinetics1,2: The precise mechanism by which trifarotene (FIGURE 1) reduces acne remains unclear. The drug, a terphenyl acid retinoid derivative, functions as an agonist of retinoic acid receptors (RARs) with particular activity at the gamma subtype of RAR, the most common RAR found in the skin. Stimulation of RARs results in modulation of target genes, which are associated with various pathologic processes, including cell differentiation and mediation of inflammation.

There is very little systemic absorption of trifarotene. The minor fraction of drug that is absorbed is highly bound by plasma proteins, metabolized by CYP2C9, CYP3A4, and CYP2C8, and excreted in the feces with a half-life of 2 to 9 hours.
Adverse Reactions and Drug Interactions1,2: In clinical trials, trifarotene was well tolerated when used on the face, back, shoulders, and chest. The most common adverse reactions (incidence >1%) included application-site irritation, pruritus, and sunburn. Patients using this drug should minimize exposure to sunlight and sunlamps and should use sunscreen and protective clothing over treated areas when exposure cannot be avoided. Depending on the severity of the skin irritation, the frequency of use of the drug should be reduced, suspended, or discontinued. Trifarotene should not be applied to cuts, abrasions, or eczematous or sunburned skin, and the use of waxing as a depilatory method should be avoided on skin treated with this drug.
Although there are case reports of major birth defects associated with the use of oral retinoids in pregnant women, data from clinical trials of trifarotene cream use in pregnancy have not revealed a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. No data exist on the presence of trifarotene in human milk, effects on breastfed infants, or effects on milk production. Based on in vitro studies, trifarotene is not expected to inhibit drug transporters or the primary cytochrome enzymes involved in drug metabolism. Therefore, topical application is not expected to pose a significant drug-interaction risk or affect the circulating concentrations of oral hormonal contraceptives containing ethinyl estradiol and levonorgestrel.
Dosage and Administration1,2: Trifarotene is supplied as a 0.005% cream in a 30-, 45- or 75-g pump for topical administration only. A thin layer of cream should be applied to the clean and dry affected areas once daily in the evening. One pump actuation should be sufficient to cover the face (i.e., forehead, cheeks, nose, and chin). Two pump actuations should be adequate to cover the upper trunk (i.e., reachable upper back, shoulders, and chest). One additional pump actuation may be used for the middle and lower back, if acne is present there. Moisturizer use is recommended as frequently as needed, beginning with initiation of treatment. Contact with the eyes, lips, paranasal creases, and mucous membranes should be avoided. The manufacturer offers a patient savings card program, Galderma CareConnect, to enable easy access to the drug.
Lemborexant (Dayvigo, Eisai)
Indication and Clinical Profile3,4: Lemborexant is a Schedule IV drug indicated specifically for treating insomnia in adults. Insomnia disorder, which is characterized by difficulty falling asleep and/or staying asleep despite adequate opportunity to sleep, can lead to a variety of secondary daytime health consequences, such as fatigue, difficulty concentrating, and irritability. Insomnia, which affects approximately 30% of the adult population worldwide, is more prevalent in women and in elderly persons.
FDA approval was based on two pivotal phase III studies (SUNRISE 1 and SUNRISE 2) in approximately 2,000 adult patients with insomnia. SUNRISE 1 was a short-term (1-month) placebo-controlled, multicenter trial in women aged 55 years and older and men aged 65 years and older who met Diagnostic and Statistical Manual of Mental Health Disorders, Fifth Edition (DSM-5), criteria for insomnia disorder. Patients were randomized to placebo (n = 208), lemborexant 5 mg (n = 266) or 10 mg (n = 269), or active comparator (n = 263) once nightly. The primary efficacy endpoint was the mean change in latency to persistent sleep (LPS). LPS was defined as the number of minutes from lights off to the first 10 consecutive minutes of nonwakefulness from baseline to end of treatment (day 29/30), as measured by overnight polysomnography monitoring. In this study, lemborexant 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure (LPS) compared with placebo. Lemborexant 5 mg and 10 mg also demonstrated statistically significant improvements in subjective sleep efficiency (sSE) and subjective sleep onset and maintenance (sWASO). sSE was considered the proportion of time spent asleep during time in bed, and sWASO was defined as minutes of wake from onset of persistent sleep until lights on.
SUNRISE 2 was a long-term (6-month) randomized, double-blind, placebo-controlled, multicenter trial in subjects aged 18 years or older who met DSM-5 criteria for insomnia disorder. Subjects were randomized to placebo (n = 325), lemborexant 5 mg (n = 323), or lemborexant 10 mg (n = 323) once nightly. The primary efficacy endpoint was the mean change from baseline to end of treatment at 6 months for subject-reported sleep onset latency (sSOL). sSOL was defined as estimated minutes from the time that the subject attempted to sleep until falling asleep. In this study, lemborexant 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure (sSOL) compared with placebo. Lemborexant 5 mg and 10 mg also showed statistically significant superiority in sSE and sWASO.
Pharmacology and Pharmacokinetics3,4: Lemborexant (FIGURE 2) is a (3-fluorophenyl)-N-(5-fluoropyridin-2-yl) cyclopropanecarboxamide analogue designed to function as a competitive antagonist of orexin-1 and orexin-2 receptors (OX1R and OX2R, respectively). The orexin neuropeptide signaling system plays a role in wakefulness. In individuals with sleep-wake disorders, orexin signaling may be dysregulated. Blocking the binding of wake-promoting neuropeptides orexin A and orexin B to OX1R and OX2R is thought to suppress wake drive.

Lemborexant is well absorbed, producing peak concentrations in 1 to 3 hours (time to maximum concentration [Tmax]). Administration with a high-fat and high-calorie meal increases Tmax by up to 2 hours. Protein binding of lemborexant is approximately 94%, and the volume of distribution of lemborexant is very large (1,970 L). Lemborexant is metabolized primarily by CYP3A4, and to a lesser extent by CYP3A5; therefore, the potential exists for CYP drug interactions. The major circulating metabolite is M10. The drug is excreted in both the feces (57%) and the urine (29%). The effective half-life for lemborexant is 17 hours for 5 mg and 19 hours for 10 mg, respectively.
Adverse Reactions and Drug Interactions3,4: The most common adverse reaction (as reported in 5% or more of patients treated with lemborexant, and at least twice the rate of placebo) in both clinical trials was somnolence, and it appears to be dose-dependent. Complex sleep behaviors including sleep-walking, sleep-driving, and engaging in other activities while not fully awake may occur; if they do, the drug should be discontinued immediately. Furthermore, patients taking the 10-mg dose of lemborexant should be cautioned against next-day driving and other activities requiring complete mental alertness. Worsening of depression or suicidal thinking may also occur; therefore, the lowest number of tablets feasible to avoid intentional overdosage should be used. Lemborexant is contraindicated in patients with narcolepsy.
No data are available on lemborexant use in pregnant women in terms of drug-associated risks of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Also, there are no data on the presence of lemborexant in human milk, effects on the breastfed infant, or effects on milk production. In rodent reproduction studies, oral administration of lemborexant during the period of organogenesis caused toxicities only at high multiples of the human exposure at the maximum recommended human dose. Because lemborexant and its metabolites were found to be present in the milk of lactating rodents, the developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for the drug and any potential adverse effects on the breastfed infant.
Lemborexant is a CYP3A4 substrate, and coadministration with strong or moderate CYP3A4 inhibitors and strong or moderate CYP3A4 inducers should be avoided. If it is coadministered with a weak CYP3A inhibitor, a maximum dosage of 5 mg is recommended.
Dosage and Administration3,4: Lemborexant is supplied as 5- and 10-mg tablets for oral administration. The recommended dosage is 5 mg taken no more than once per night, immediately before bedtime, with at least 7 hours remaining before the planned time of awakening. The dose may be increased to the maximum of 10 mg based on clinical response and tolerability. Time to sleep onset may be delayed if the drug is taken with or soon after a meal. No dose adjustment is required in patients with mild, moderate, or severe renal impairment. If lemborexant is used concurrently with a weak CYP3A inhibitor, a maximum of 5 mg is recommended. In patients with moderate hepatic impairment, the maximum recommended dosage is 5 mg no more than once per night. The drug is not recommended in severe hepatic impairment.
Lactitol (Pizensy, Braintree)
Indication and Clinical Profile5,6: Lactitol is specifically indicated for the treatment of chronic idiopathic constipation (CIC) in adults. CIC is a complex gastrointestinal (GI) disorder with no identifiable cause that is characterized by symptoms ranging from less than three bowel movements per week to hard-to-pass or incomplete bowel movements. This disorder affects approximately 33 million Americans and an estimated 14% of the worldwide population. Nearly 4.7 million brand prescriptions were written for CIC-alleviating products over the past year. Lactitol is different from other CIC prescriptions in that rather than a solid-form oral dose, this product is a powder for reconstitution that offers the ability to mix the medication with the patient’s preferred fluid, including water, juice, coffee, tea, and soda. Lactitol also allows patients the freedom of choosing what time of day to take it, as well as enabling them to adjust the dosage.
FDA approval was based on results of three phase III controlled clinical trials (i.e., 1) a 6-month study; 2) a 12-week, blinded, head-to-head study against lubiprostone; and 3) a 12-month, open-label, chronic-use study) involving more than 1,400 patients. The primary efficacy analysis was based on the first 12 weeks of the 6-month treatment period among 594 patients, with 291 receiving lactitol and 303 receiving placebo. Significantly more patients using lactitol responded to treatment, compared with placebo (25% vs. 13%). A responder was defined as a patient who had at least three complete spontaneous bowel movements (CSBMs) in a given week and an increase of at least one CSBM from baseline in the same week for at least 9 weeks of the first 12-week treatment period and at least three of the last 4 weeks. During clinical trials, approximately 25% of participants had the option of reducing the dosage. A significant number of patients who received lactitol in clinical trials (25%) were aged 65 years or older and 7% were aged 75 years or older, but no overall differences in safety or efficacy were observed between geriatric patients and younger patients.
Pharmacology and Pharmacokinetics5,6: Lactitol (FIGURE 3) is a simple monosaccharide sugar alcohol that acts as an osmotic laxative. This osmotic effect draws water into the small intestine, creating a hyperosmotic environment that loosens stools and ultimately facilitates bowel movements. Lactitol is minimally absorbed systemically following oral administration; therefore, there is no significant distribution, metabolism, and excretion profile. Unabsorbed lactitol appears to be degraded into organic acids in the colon and is minimally excreted in the feces.

Adverse Reactions and Drug Interactions5,6: The most frequent adverse effect (AE) of lactitol reported during clinical trials involved upper respiratory tract infections (9% in the treatment groups vs. 6% in the placebo group). Less frequent AEs included flatulence and diarrhea, abdominal distention, increased blood creatinine phosphokinase, and increased blood pressure. Overall, discontinuation rates for this drug were low in clinical trials (4%).
Lactitol is contraindicated in patients with mechanical GI obstruction and galactosemia. The drug is minimally absorbed systemically following oral administration, but it is unknown whether maternal use will result in fetal exposure to the drug in utero or during breastfeeding. Owing to lack of absorption, no systemic metabolic or excretion drug interactions are anticipated.
Dosage and Administration5,6: Lactitol is supplied as an off-white crystalline powder for reconstitution as an oral solution in 10-g unit-dose packets and 280-g and 560-g multidose bottles. The multidose bottles are equipped with a measuring cap marked to contain 10 g of powder when filled to the top of the white section in the cap. The recommended daily dosage is 20 g mixed with 4 oz to 8 oz of water, juice, coffee, tea, or soda, preferably with a meal. The dosage should be reduced to 10 g daily in patients who have persistent loose stools. Since lactitol can reduce the absorption of concomitantly administered oral medications, it may be administered at least 2 hours before or 2 hours after other drugs.
Ubrogepant (Ubrelvy, Allergan)
Indication and Clinical Profile7,8: Ubrogepant is specifically indicated for acute treatment of migraine with or without aura in adults. It is the first drug in the class of oral calcitonin gene-related peptide (CGRP) receptor antagonists to be approved for acute treatment of migraine, but it is not indicated for preventive treatment of migraine. Nearly 40 million people in the United States suffer from migraine, which the World Health Organization classifies as one of the 10 most disabling illnesses. Migraine is characterized by debilitating attacks lasting 4 to 72 hours that involve multiple symptoms, including pulsating headaches of moderate-to-severe pain intensity that may be associated with nausea or vomiting, sensitivity to sound, and/or sensitivity to light. There is a significant unmet need for new acute treatments, as more than 90% of migraineurs are unable to work or function normally during an attack.
FDA approval was based on two randomized, double-blind, placebo-controlled trials. In these studies, 1,439 adult patients with a history of migraine, with and without aura, received the approved doses of ubrogepant to treat an ongoing migraine. In both studies, the percentage of patients achieving pain freedom 2 hours after treatment (defined as a reduction in headache severity from moderate or severe pain to no pain) and the percentage of those whose most bothersome symptom (nausea, light sensitivity, or sound sensitivity) stopped 2 hours after treatment were significantly greater ain patients who received ubrogepant, at all doses, compared with those who received placebo. Patients were allowed to take their usual acute migraine treatments at least 2 hours after taking ubrogepant, and 23% of patients were taking a preventive medication for migraine.
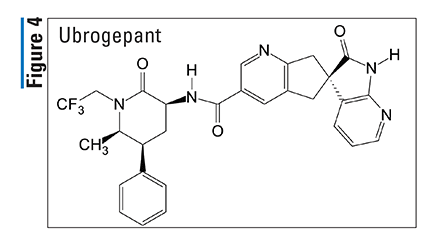
Pharmacology and Pharmacokinetics7,8: Ubrogepant (FIGURE 4) is a tetrahydrospiro[cyclopenta-[b]pyridine-6,3ʹ-pyrrolo[2,3b]pyridine]-3-carboxamide specially designed to function as a CGRP receptor antagonist. The activity of the neuropeptide CGRP acts at multiple sites along the trigeminovascular pathway, causing vasodilation and inflammation and amplifying and perpetuating migraine pain. Small-molecule CGRP receptor antagonists represent a novel class of drugs for the treatment of migraine. This unique mode of action offers a potential alternative to current agents, particularly for patients who have contraindications to the use of triptans or who have a poor response to triptans or are intolerant to them.
Ubrogepant is well absorbed from the GI tract, providing peak plasma concentrations at approximately 1.5 hours. Coadministration of a high-fat meal delays the time to maximum ubrogepant plasma concentration by 2 hours and results in a 22% reduction in maximum plasma concentration with no change in AUC. Ubrogepant displays dose-proportional pharmacokinetics within the recommended dosing range. Plasma protein binding is 87%, and the mean apparent central volume of distribution is approximately 350 L. Ubrogepant is eliminated mainly through CYP3A4 metabolism followed by formation of an inactive glucuronide conjugate. The parent drug and its glucuronide metabolites are the most prevalent circulating components in human plasma. The elimination half-life of ubrogepant is approximately 5 to 7 hours, with a mean apparent oral clearance of approximately 87 L per hour. Ubrogepant is a substrate of breast cancer resistance protein (BCRP) and P-glycoprotein (Pgp) efflux transporters and is excreted mostly via the biliary/fecal route. In patients with preexisting mild (Child-Pugh Class A), moderate (Child-Pugh Class B), or severe (Child-Pugh Class C) hepatic impairment, ubrogepant exposure is increased by 7%, 50%, and 115%, respectively. No dosage adjustment for ubrogepant is recommended for patients with mild or moderate hepatic impairment; however, adjustment is recommended for patients with severe hepatic impairment. Since the renal route of elimination plays a minor role in the clearance of ubrogepant, no dosage adjustment is recommended for patients with mild or moderate renal impairment. Adjustment is recommended for patients with severe renal impairment (creatinine clearance [CrCl] 15-29 mL/min), and the use of ubrogepant should be avoided in patients with end-stage renal disease (ESRD) (CrCl <15 mL/min). In pharmacokinetic studies, no clinically significant differences were observed between elderly and younger subjects.
Adverse Reactions and Drug Interactions7,8: The most common side effects reported in clinical trials were nausea, somnolence, and dry mouth. No adequate data exist on the developmental risk associated with ubrogepant use in pregnant women. In animal studies, adverse effects on embryofetal development were observed following administration of ubrogepant during pregnancy and lactation. Therefore, a patient should notify her healthcare provider if she becomes pregnant, plans to become pregnant, is breastfeeding, or plans to breastfeed during treatment.
Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors. Also, coadministration of ubrogepant with strong CYP3A4 inducers should be avoided because their use results in a reduction of ubrogepant exposure. Dosage adjustments are recommended for patients who are taking moderate or weak CYP3A4 inhibitors or inhibitors of the hepatic transporters BCRP and Pgp.
Dosage and Administration7,8: Ubrogepant is available in 50- and 100-mg tablets. The recommended dosage is 50 mg or 100 mg taken orally with or without food. If needed, a second dose may be taken at least 2 hours after the initial dose. The maximum dose in a 24-hour period is 200 mg. The safety of treating more than eight migraines in a 30-day period has not been established. In patients with severe hepatic or severe renal impairment, the recommended dose is 50 mg; if needed, a second 50-mg dose may be taken at least 2 hours after the initial dose. Ubrogepant should be avoided in patients with ESRD. For patients taking concomitant moderate or weak CYP3A4 inhibitors or BCRP and/or Pgp inhibitors, the initial dose should be limited to 50 mg. If a second dose is needed with a weak CYP3A4 inhibitor or a Pgp or BCRP inhibitor, the second dose should be limited to 50 mg. The use of a second 50-mg dose should be avoided within 24 hours in patients who are taking a moderate CYP3A4 inhibitor.
Rimegepant (Nurtec ODT, Biohaven)
Indication and Clinical Profile9,10: Rimegepant is specifically indicated for acute treatment of migraine with or without aura in adults. It is not indicated for preventive treatment of migraine. This drug is the first and only calcitonin gene-related peptide (CGRP) receptor antagonist available in a fast-acting orally disintegrating tablet (ODT).
FDA approval is based on results of a pivotal phase III clinical trial (Study 303) and a long-term, open-label safety study (Study 201). In the phase III trial, rimegepant ODT achieved statistical significance on the regulatory coprimary endpoints of pain freedom and freedom from most bothersome symptom (MBS) at 2 hours post dose compared with placebo. Rimegepant ODT also demonstrated statistical superiority at 1 hour for pain relief (reduction of moderate or severe pain to no pain or mild pain) and return to normal function. The benefits of pain freedom, pain relief, return to normal function, and freedom from MBS were sustained up to 48 hours for many patients. These benefits were seen with only a single dose of rimegepant ODT, and 86% of patients treated with rimegepant did not require rescue medication within 24 hours post dose.
Pharmacology and Pharmacokinetics9,10: Rimegepant (FIGURE 5) is an imidazo[4,5-b]pyridin-1-yl)-1-piperidinecarboxylate derivative designed to function as a CGRP receptor reversible antagonist, similar to ubrogepant and other drugs of this class.

Rimegepant disperses almost instantly in the mouth without the need for water, offering migraine patients a convenient, discreet way to take the medication. Following oral administration, rimegepant produces a maximum concentration at 1.5 hours, with an absolute oral bioavailability of 64%. Administration with a high-fat meal delays time to maximum concentration by 1 hour and appears to result in a 40% to 50% reduction in maximum plasma concentration and a 32% to 38% reduction in AUC. The steady-state volume of distribution of rimegepant is 120 L, and plasma protein binding is approximately 96%. Rimegepant undergoes some metabolism, primarily by CYP3A4 and, to a lesser extent, CYP2C9. Following oral administration, most of the drug (78%) is excreted in the feces with 24% in the urine. Unchanged drug is the major single component in excreted feces (42%) and urine (51%). The elimination half-life of rimegepant is approximately 11 hours in healthy subjects.
Adverse Reactions and Drug Interactions9,10: The most common adverse effect associated with rimegepant in clinical trials was nausea. The drug is contraindicated in patients with a history of hypersensitivity to rimegepant or to any of its components. Hypersensitivity reactions with dyspnea and severe rash, including delayed serious hypersensitivity days after administration, occurred in less than 1% of subjects taking rimegepant in clinical studies. If a serious hypersensitivity reaction occurs, rimegepant should be discontinued immediately and appropriate therapy initiated. No adequate data exist on the developmental risk associated with the use of rimegepant during pregnancy and breastfeeding.
Rimegepant is a substrate of CYP3A4 and P-glycoprotein (Pgp) and BCRP efflux transporters. Therefore, its use should be avoided in patients receiving concurrent therapy with a strong CYP3A4 inhibitor, a strong-to-moderate CYP3A4 inducer, and inhibitors of Pgp or BCRP. A second dose of rimegepant should be avoided within 48 hours if coadministered with a moderate CYP3A4 inhibitor.
Dosage and Administration9,10: Rimegepant is supplied as a tablet for sublingual or oral administration. The recommended dosage is 75 mg taken orally. The maximum dosage in a 24-hour period is 75 mg. The safety of treating more than 15 migraines in a 30-day period has not been established. The use of rimegepant in patients with severe hepatic impairment or end-stage renal disease should be avoided.
REFERENCES
1. Aklief (trifarotene) package insert. Fort Worth, TX: Galderma Laboratories, LP; November 2019.
2. Tan J, Thiboutot D, Popp G, et al. Randomized phase 3 evaluation of trifarotene 50 µg/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019;80(6):1691-1699.
3. Dayvigo (lemborexant) package insert. Woodcliff Lake, NJ: Eisai Inc; December 2019.
4. Cappuccio FP, D’Elia L, Strazzullo P, Miller MA. Sleep duration and all-cause mortality: a systematic review and meta-analysis of prospective studies. Sleep. 2010;33(5):585-592.
5. Pizensy (lactitol) package insert. Braintree, MA: Braintree Laboratories, Inc; February 2020.
6. Miller LE, Tennilä J, Ouwehand AC. Efficacy and tolerance of lactitol supplementation for adult constipation: a systematic review and meta-analysis. Clin Exp Gastroenterol. 2014;7:241-248.
7. Ubrelvy (ubrogepant) package insert. Madison, NJ: Allergan USA, Inc; December 2019.
8. Dodick DW, Lipton RB, Ailani J, et al. Ubrogepant for the treatment of migraine. N Engl J Med. 2019;381(23):2230-2241.
9. Nurtec (rimegepant) package insert. New Haven, CT: Biohaven Pharmaceuticals, Inc; February 2020.
10. Croop R, Ivans A, Stock D, et al. A phase 1 study to evaluate the bioequivalence of oral tablet and orally dissolving tablet formulations of rimegepant, a small molecule CGRP receptor antagonist. Poster presented at: London, UK: 17th Biennial Migraine Trust International Symposium; September 6-9, 2018; London, UK.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


