US Pharm.
2008;33(1):HS-3-HS-19.
With the increasing
development of newer and more complex drug compounds, pharmacists are being
asked to bear much of the burden for detecting, preventing, and resolving
adverse drug reactions and potentially serious drug interactions. Factors that
contribute to drug interactions can be easily identified, but knowing how to
prevent interactions is far more difficult. Drug interactions can mislead
clinicians into misinterpreting these effects as unrelated to adverse
drugñdrug interactions. The following scenario illustrates the quandary in
which pharmacists find themselves in evaluating drug interactions to determine
safe use: A physician contacts a pharmacist and asks which selective serotonin
reuptake inhibitor (SSRI) can be used safely in a patient taking multiple
medications (i.e., warfarin, thiazide diuretic, beta blocker, and codeine).
Since there is a high potential for clinically significant drug interactions
with these drugs, advising this clinician will require some thought and
research.
National concern about drug
interactions with cytochrome P (CYP)-450 enzymes was heightened when fatal
cardiac arrhythmias were suspected to be connected to enzymatic interactions
between terfenadine and erythromycin or ketoconazole.1,2 As a
result, terfenadine was withdrawn from the market. Later, mibefradil and
cisapride were withdrawn due to their high potential for inhibiting certain
CYP enzymes and causing fatal cardiac arrhythmias when combined with certain
CYP-enzyme inhibitors. As a consequence, some drugs marketed in the last two
decades--especially antidepressants--that have been associated with serious
drugñdrug interactions have been subject to careful scientific examination.
Because chronic illnesses,
especially depression, require extended periods of treatment, the probability
of co-administration of additional medications is high. The increased
prevalence of depression in both the young and the elderly populations has led
to the addition of antidepressants to complex medication regimens. In 2006,
antidepressant utilization in the United States was extensive. Three of the
top 15 and five of the top 50 brand drugs dispensed by pharmacies were
antidepressants, as were 10 of the top 200 generic drugs.3
Drug interactions are of
concern because the outcome of concurrent drug administration is diminished
therapeutic efficacy or increased toxicity of one or more of the administered
compounds. Mechanisms of drug interactions are usually divided into two major
categories, pharmacokinetic and pharmacodynamic. Pharmacokinetic interactions
consist of changes in the absorption, distribution, metabolism, or excretion
of a drug or its metabolites, or the quantity of active drug that reaches its
site of action, after the addition of another chemical agent.
Metabolically-based drug interactions are the most frequent interaction
encountered in clinical practice. Pharmacodynamic interactions occur when two
drugs act at the same or interrelated receptor sites, resulting in additive,
synergistic, or antagonistic effects. The purpose of this article is to give
pharmacists an overview of metabolic drugñdrug interactions involving SSRIs.
Drug Metabolism and
Overview of the CYP System
Psychotropic drugs,
including many antidepressants, are usually lipophilic and are extensively
metabolized in the liver through phase I oxidative reactions followed by phase
II glucuronide conjugation. Most pharmacokinetic interactions with
psychotropic drugs occur at the metabolic level and primarily involve the CYP
mono-oxygenases. In some instances, the metabolite of the parent compound has
a greater inhibitory effect on the metabolizing CYP isoenzyme(s). Thus, the
potential for drug interactions may be greater in clinical practice, where
patients may receive higher initial doses or receive doses that are titrated
to reach steady-state levels.4
Enzymes of the CYP system are
classified into families, subfamilies, and isoenzymes based on similarities in
the sequences of their amino acids.5,6 CYP enzymes are responsible
for the oxidative metabolism of xenobiotics (drugs and other exogenous
chemicals), as well as many endogenous compounds such as prostaglandins, fatty
acids, and steroids. The first Arabic number designates the enzyme family, the
capital letter indicates the subfamily, and the second number designates
individual isoenzymes. The major CYP enzymes involved in drug metabolism in
humans belong to families 1, 2, and 3, the specific isoforms being CYP-1A2,
CYP-2C9, CYP-2C19, CYP-2D6, and CYP-3A4. (Due to their identical structure and
enzymatic action, CYP-3A3 and CYP-4 are often combined and referred to as
CYP-3A4.) Each CYP isoform is a specific gene product and possesses a
characteristic broad spectrum of substrate specificity. The activity of these
isoenzymes is genetically determined and is greatly influenced by
environmental factors, such as concomitant administration of other drugs.
Drug interactions involving
CYP isoforms generally result from one of two processes: enzyme inhibition and
enzyme induction. Enzyme inhibition usually involves competition with another
drug for the enzyme-binding site. Drug-induced inhibition of CYP enzymes is
usually due to competitive binding at enzyme-binding sites, and it generally
occurs within a few hours.7-9 The magnitude of the inhibition is a
function of the plasma concentration of the inhibiting agent. Thus, the
half-life of the inhibitor drug will determine how long it must be
administered before the full inhibitory effect on CYP enzymes is achieved and,
conversely, how long after its discontinuation the inhibition phase will
endure.
Enzyme induction occurs when a
drug stimulates the synthesis of more enzyme protein, enhancing the enzyme's
metabolizing capacity. Induction of the gene responsible for the production of
the enzyme increases its rate of synthesis of the drug, thus increasing the
cellular content and activity of the induced CYP enzymes.10-12
Since enzyme induction generally involves protein synthesis, there is a time
delay in both the onset and the offset relative to starting and stopping the
inducing agent. Therefore, the full effect of the inducer may not be evident
for several weeks after the inducer drug has been started. The resulting
effect will take a similar period of time to fully dissipate after the inducer
agent has been discontinued and the rate of enzyme production has returned to
baseline.
SSRIs
Currently, five
types of SSRIs are marketed in the United States: fluoxetine, fluvoxamine,
paroxetine, sertraline, and citalopram. These drugs are subject to extensive
oxidative metabolism in the liver. Because these antidepressants have a wide
therapeutic index, inhibition or induction of their metabolism is unlikely to
be of great concern. However, SSRIs may cause a clinically relevant inhibition
of CYP enzymes, and care must be exercised when an SSRI is being added to a
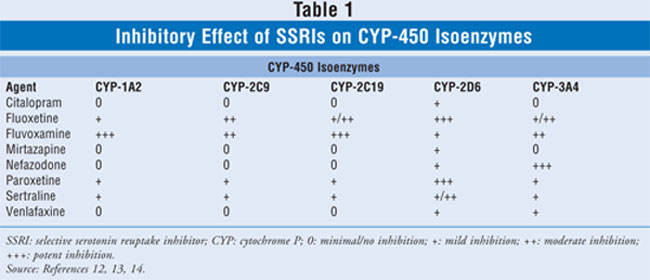
multidrug regimen. As shown in TABLE 1, SSRIs differ considerably in
their ability to inhibit individual CYP enzymes. This may help guide selection
of an appropriate compound for the individual patient.12,13 The
inhibitory effect on CYP enzymes is concentration-dependent; the potential for
drug interactions with citalopram and paroxetine is higher in the elderly
because the elimination of these drugs may be affected by age. This is
especially true with drugs such as fluoxetine, which exhibits nonlinear
kinetics.
Fluoxetine
Fluoxetine is
marketed as a racemic mixture of two enantiomers.14 The major
metabolic pathway of fluoxetine is N-demethylation to form the active
metabolite norfluoxetine. In vivo studies have indicated that CYP-2D6 is the
major isoform responsible for the N-demethylation of fluoxetine. In
vitro evidence, however, suggests that other isoenzymes, including CYP-2C9,
CYP-2C19, and CYP-3A4, also may contribute to this reaction. Fluoxetine and
its metabolite norfluoxetine have important inhibitory effects on CYP enzymes
in vitro. They were found to inhibit CYP-2D6 markedly, CYP-2C9 moderately, and
CYP-2C19 and CYP-3A4 mildly to moderately.
Fluoxetine follows nonlinear
kinetics, and its plasma concentrations increase to a greater extent than the
increase in drug dosages would predict. When fluoxetine is taken routinely, it
takes about one month for it to reach a steady-state level in the blood and
cause a drug interaction. Due to the long elimination half-lives of fluoxetine
(one to four days) and norfluoxetine (seven to five days), inhibition of CYP
enzymes may persist for up to six weeks after discontinuation of the
antidepressant, a situation that complicates patient management.
Drug Interactions:
Fluoxetine 20-60 mg/day may cause a two- to fourfold increase in plasma
concentrations of desipramine, possibly associated with signs of toxicity
including decreased energy, psychomotor retardation, sedation, dry mouth, and
memory loss.15,16 The mechanism of this interaction may be
attributed to the potent inhibitory effect of fluoxetine and norfluoxetine on
the CYP-2D6ñmediated hydroxylation of tricyclic antidepressants (TCAs). When
given in combination with the heterocyclic antidepressant trazodone,
fluoxetine was found to produce a significant elevation in plasma levels of
both trazodone and its metabolite metachlorophenylpiperazine (mCPP).15,17
This reaction is probably caused by the inhibition of CYP-2D6 and CYP-3A4 in
the metabolism of trazodone and by CYP-2D6 inhibition of mCPP metabolism.
Fluoxetine has been reported
to produce a remarkable increase in plasma concentrations of traditional
antipsychotics such as haloperidol and fluphenazine, metabolized at least in
part by CYP-2D6, possibly leading to adverse central nervous system (CNS)
effects such as extrapyramidal symptoms and impaired psychomotor performance.
15,18 Fluoxetine also may interfere with the elimination of some new
atypical antipsychotics. With the coadministration of clozapine and fluoxetine
20 mg/day, one report showed an increase of 50% to 100% in plasma
concentrations of cloza!= pine, an atypical antipsychotic metabolized by
CYP-1A2 and, to a lesser extent, by CYP-3A4, CYP-2D6, and CYP-2C19.15,19
Concomitant treatment with fluoxetine 20 mg/day in psychotic patients
stabilized on risperidone, an antipsychotic whose metabolism is largely
dependent on CYP-2D6 and CYP-3A4, was associated with a mean fourfold increase
in plasma concentrations of risperidone.20 As a consequence, the
active fraction of risperidone (sum of plasma concentrations of risperidone
and its active metabolite) increased by 76% over pretreatment. Patients
reported the occurrence of akathisia and parkinsonian symptoms requiring
anticholinergic medication.21
Fluoxetine may impair the
elimination of some benzodiazepines such as diazepam and alprazolam through
inhibition of the major CYP isoforms mediating their metabolism, in particular
CYP-2C19 (diazepam) and CYP-3A4 (diazepam, alprazolam).15,21,22 As
benzodiazepines have a wide therapeutic index, however, the clinical
significance of these interactions is probably limited. Fluoxetine also may
impair the elimination of phenytoin, as documented by many case reports of
toxic phenytoin concentrations occurring shortly after the addition of
fluoxetine.15,23 This interaction is probably explained by the
moderate inhibitory effect of fluoxetine on the CYP-2C9ñmediated metabolism of
phenytoin.24
A clinically significant
interaction can occur between fluoxetine and warfarin, resulting in enhanced
anticoagulant activity and a subsequent risk of hemorrhagic complications, as
well as marked elevation of the international normalized ratio (INR) and
prolongation of prothrombin time, in patients stabilized on warfarin.15,25
The inhibitory effect of fluoxetine on CYP-2C9ñmediated metabolism of active
S-warfarin is the most likely explanation for this potentially serious
drug interaction. Since warfarin is a racemic mixture with an active S
-enantiomer and a less active R-enantiomer, S-warfarin is
metabolized by CYP-2C9, while R-warfarin is metabolized by CYP-1A2 and,
to a lesser extent, CYP-2C19 and CYP-3A4. In addition to being metabolized by
these isoenzymes, R-warfarin inhibits CYP-2C9 activity, thus increasing
the effect of active S-warfarin.26
There are some reports of
potentially dangerous interactions between fluoxetine and certain
cardiovascular agents. Inhibition of the oxidative metabolism of beta blockers
(metoprolol, propranolol), which is partly mediated by CYP-2D6, may explain
the occurrence of severe bradycardia or heart block in patients after
coadministration of fluoxetine.15,27 The combination of fluoxetine
and the calcium channel blockers nifedipine and verapamil has been reported to
be associated with signs of toxicity such as edema, nausea, and flushing that
disappeared when the dose of the calcium channel antagonists was reduced.
15,28 Inhibition of CYP-3A4ñmediated metabolism of verapamil and
nifedi!= pine by fluoxetine and its metabolite norfluoxetine may explain the
occurrence of this interaction.
Fluvoxamine
The major metabolic
pathways of fluvoxamine are oxidative demethylation and oxidative deamination
by CYP-2D6 and CYP-1A2.15,29 Fluvoxamine interacts with several CYP
isoenzymes. It is a potent inhibitor of CYP-1A2 and CYP-2C19 and a moderate
inhibitor of CYP-2C9 and CYP-3A4; it affects CYP-2D6 activity only slightly.
30 As a result of this nonselective inhibition of various CYP
isoenzymes, fluvoxamine has a high potential for metabolic drug interactions.
Drug Interactions:
Fluvoxamine may increase the plasma concentrations of certain
antidepressants. Fluvoxamine affects predominantly the demethylation pathways
of TCAs through inhibition of CYP-2C19 and, to a lesser extent, CYP-1A2 and
CYP-3A4. Accordingly, plasma levels of the tertiary amines amitriptyline,
imipramine, and clomipramine have been reported to increase by up to fourfold
during coadministration with fluvoxamine 100 mg/day, possibly leading to toxic
effects, while concentrations of the secondary amine desipramine were only
slightly modified.31 A recent report documented that the addition
of fluvoxamine 50-100 mg/day caused a three- to fourfold increase in plasma
concentrations of mirtazapine, a new antidepressant metabolized mainly by
CYP-1A2, CYP-2D6, and CYP-3A4.32
Fluvoxamine may interfere with
the biotransformation of various antipsychotics. The addition of fluvoxamine
50-300 mg/day to haloperidol maintenance therapy in patients with
schizophrenia resulted in a 1.8- to 4.2-fold increase in serum haloperidol
concentrations.33 This interaction is likely explained by the
inhibitory effect of fluvoxamine on CYP-1A2 and CYP-3A4, which are involved in
the metabolism of haloperidol.
Clinically relevant metabolic
interactions may occur between fluvoxamine and the atypical antipsychotics
clozapine and olanzapine. Researchers have clearly documented that fluvoxamine
may increase plasma clozapine concentration up to five- to 10-fold, possibly
resulting in toxic effects.34 Therefore, the combination of
fluvoxamine and clozapine must be carefully monitored, and the use of low
doses of both compounds is advisable. This interaction is attributed not only
to inhibition of CYP-1A2, the major enzyme responsible for clozapine
metabolism, but also to additional inhibitory effects of fluvoxamine on
CYP-2C19 and CYP-3A4.
Fluvoxamine may elevate plasma
levels of olanza!= pine by approximately twofold.35 The potent
inhibitory effect of fluvoxamine on CYP-1A2, one of the major isoforms
responsible for olanzapine biotransformation, provides a rational explanation
for this interaction. Fluvoxamine also has been reported to decrease the
metabolic clearance of some benzodiazepines, including alprazolam, which is
metabolized primarily by CYP-3A4, and diazepam, which is substrate for both
CYP-2C19 and CYP-3A4.36,37
Potentially dangerous
consequences resulting from the combined use of fluvoxamine and theophylline
have been documented.38 Concomitant treatment with fluvoxamine may
cause a marked elevation in plasma theophylline levels associated with signs
of theophylline toxicity, including ventricular tachycardia, anorexia, nausea,
and seizures. This interaction is presumably mediated by the inhibitory effect
of fluvoxamine on the activity of CYP-1A2, which is the main isoenzyme
involved in theophylline metabolism. Theophylline toxicity is a serious,
sometimes fatal, medical condition, so fluvoxamine should be avoided in
patients taking theophylline.
A potentially dangerous
interaction may occur between fluvoxamine and warfarin. The addition of
fluvoxa!= mine for two weeks to a stable regimen of warfarin produced a 65%
increase in plasma warfarin concentration and a significant prolongation of
prothrombin time.15 In one report, the addition of a low dose of
fluvoxamine in the case of an elderly woman with atrial fibrillation that was
stabilized on warfarin resulted in a marked elevation of her INR that
persisted for two weeks after the antidepressant was stopped.39 The
mechanism of this interaction is particularly complex. In fact, fluvoxamine
may directly increase plasma levels of S-warfarin through its moderate
inhibitory effect on CYP-2C9. In addition, fluvoxamine, a strong inhibitor of
CYP-1A2, is expected to elevate R-warfarin levels, which in turn would
reduce CYP-2C9 activity and thus increase the effect of the active S
-warfarin.25
In a study of healthy
volunteers, coadministration of fluvoxamine 100 mg/day with propranolol 160
mg/day resulted in a fivefold increase in plasma propranolol concentrations
that was associated with a slight potentiation of the propranolol-induced
reductions in heart rate and exercise diastolic blood pressure.15
This effect is likely to be the consequence of an inhibitory effect of
fluvoxamine on CYP-1A2 and CYP-2C19, the major isoforms involved in the
biotransformation of this beta blocker.
In addicted patients on
maintenance treatment with methadone--a synthetic opioid predominantly
metabolized via CYP-3A4--fluvoxamine, but not fluoxetine, was found to
increase plasma concentrations of both methadone enantiomers by 30% to 50%.
40,41 As is the case with fluoxetine, fluvoxamine may cause potentially
serious interactions if it is coadministered with CYP-3A4 substrates.
Paroxetine
Among the SSRIs,
paroxetine is the most potent in vitro inhibitor of CYP-2D6, although it
affects other CYP isoforms only minimally.15,42 Paroxetine
therefore has the potential to cause clinically significant drug interactions
when coadministered with CYP-2D6 substrates. It undergoes extensive hepatic
biotransformation, including oxidative cleavage mediated by CYP enzymes, while
methylation reactions are probably mediated by catechol-O
-methyltransferase. Oxidation of paroxetine is likely catalyzed by a main
pathway mediated by CYP-2D6, whose saturation is responsible for the drug's
nonlinear kinetics (i.e., plasma concentrations increase to a greater extent
than the increase in drug dosages would predict), and by a secondary pathway
presumably mediated by CYP-3A4. This isoenzyme is usually responsible for 25%
of biotransformation, but becomes more significant at higher plasma
concentrations.43 An intermediate metabolite of paroxetine has been
shown to have inhibitory activity against CYP-2D6.
Drug Interactions:
Like fluoxetine, paroxetine may inhibit CYP-2D6ñmediated hydroxylation of
TCAs, possibly leading to adverse effects. When paroxetine was dosed at 20
mg/day and under steady-state conditions, it increased plasma concentrations
of desipramine (a substrate of CYP-2D6) from 327% to 421%.44,45
Paroxetine may impair the elimination of older and newer antipsychotics
metabolized by CYP-2D6. In a study of healthy volunteers, paroxetine was found
to cause a two- to 13-fold increase in single-dose perphenazine peak plasma
concentrations, with associated CNS effects such as sedation and
extrapyramidal symptoms.46
Paroxetine 20 mg/day given to
patients with schizophrenia produced a three- to ninefold elevation in plasma
levels of risperidone, resulting in a mean 45% increase in plasma
concentrations of the active fraction of risperidone.15 These
changes were associated with the occurrence or worsening of extrapyramidal
side effects. Other studies have reported that paroxetine may produce a
moderate elevation in plasma concentrations of clozapine.47
Sertraline
The major metabolic
pathway of sertraline is N-demethylation to form N
-desmethylsertraline, which is less potent than the parent drug as a serotonin
reuptake blocker. CYP-3A4 is the major isoform responsible for this reaction,
but other isoenzymes, including CYP-2D6, probably are involved.48
In vitro studies have documented that sertraline is a mild to moderate
inhibitor of CYP-2D6 and a weak inhibitor of the other CYP isoenzymes; this
accounts for its favorable interaction profile.15,44
Drug Interactions:
Sertraline 50 mg/day was found to cause modifications in plasma
concentrations of TCAs, but these were less pronounced compared with other
SSRIs.49 Because the inhibition of CYP-2D6 is dose-dependent,
however, significant increases in plasma concentrations of TCAs may occur when
higher dosages of sertraline are administered.50
Citalopram
Citalopram is a
racemic mixture, with its antidepressant effects attributed exclusively to the
S (+)-enantiomer. S-citalopram (escitalopram) was recently
introduced as an antidepressant.15 The major CYP isoenzymes
involved in the metabolism of citalopram are CYP-2C19 and CYP-2D6, with
didesmethylcitalopram being the final metabolite. In vitro studies have
indicated that CYP-3A4 also is involved in the N-demethylation of
citalopram. Citalopram is a weak in vitro inhibitor of CYP-2D6, and it has
weak or no effects on CYP-1A2, CYP-2C19, and CYP-3A4.51
Drug Interactions:
With respect to pharmacokinetic drug interactions, citalopram is not the
cause or the source of clinically significant drug interactions. Therefore, it
is considered the safest SSRI to use in clinical practice.
Venlafaxine
Venlafaxine, a
serotonin and noradrenaline reuptake inhibitor, is biotransformed to a major
active metabolite, O-desmethylvenlafaxine, and is in parallel with N
-desmethylvenlafaxine. In vitro and in vivo studies reported that the O
-demethylation of venlafaxine is catalyzed mainly by CYP-2D6, while CYP-3A4
is probably involved in the N-demethylation pathway.15,52 In
vitro studies demonstrated that venlafaxine is a weak inhibitor of CYP-2D6,
but is considerably less potent than paroxetine, fluoxetine, fluvoxamine, and
sertraline and does not significantly affect the activity of CYP-1A2, CYP-2C9,
and CYP-3A4. Venlafaxine has a relatively short half-life of five to 11 hours,
takes three to five days to reach steady state, and may be associated with
clinical drug interactions soon after treatment is initiated.
Drug Interactions:
Based on in vitro evidence, venlafaxine appears to have minimal effects on
the pharmacokinetics of other drugs.16,53 When venlafaxine was
dosed at 150 mg/day in healthy subjects, imipramine and desipramine clearance
was slightly reduced, leading to significant increases in their area under the
curve (AUC) of 27% and 40%, respectively. In another in vivo study,
coadministration of venlafaxine 150 mg/day, with a single 1-mg dose of
risperidone, slightly inhibited its conversion of risperidone to
9-hydroxyrisperidone (9-OH-risperidone), which is partially metabolized by
CYP-2D6. Although the exact mechanism remains uncertain, venlafaxine caused a
70% increase in the AUC of coadministered haloperidol.
Mirtazapine
Mirtazapine is the
first in a new class of antidepressants, the noradrenergic and specific
serotonergic antidepressants. Its effect appears to be related to its dual
enhancement of central noradrenergic and serotonin 5-HT1
receptorñmediated serotonergic neurotransmission. Mirtazapine is extensively
metabolized in the liver; its major metabolic routes are N
-demethylation, N-oxidation, and 8-hydroxylation. CYP-2D6 and, to a
lesser extent, CYP-3A4 are involved in the formation of hydroxymetabolites.
CYP-3A4 and CYP-1A2 catalyze the N-demethylation, while CYP-3A4 is the
major isoform involved in N-oxidation.15,54
Drug Interactions:
Mirtazapine has minimal inhibitory effects on CYP-1A2, CYP-2D6, and CYP-3A4.
Therefore, it is not expected to cause clinically significant interactions
with substrates of these isoforms.
Nefazodone
Nefazodone is a potent serotonin 5-HT
2 receptor antagonist that inhibits both serotonin and noradrenaline
reuptake. It is extensively metabolized in the liver by hydroxylation and
dealkylation, primarily via CYP-3A4.15,55 Hydroxynefazodone, the
major metabolite, displays pharmacologic activity similar to its parent drug.
Other minor metabolites include mCPP and a triazoledione derivative, both of
which are less active than nefazodone. Nefazodone has been shown in vitro to
be a potent inhibitor of CYP-3A4; it also has a weak inhibitory effect on
CYP-2D6 activity, presumably due to mCPP.
Drug Interactions:
When nefazodone 200 mg twice daily was given to healthy volunteers for seven
days, results included an increase in plasma concentrations of triazolam and
alprazolam--substrates of CYP-3A4--of 98% and 290%, respectively.15,56
The most clinically important
drug interactions with nefazodone may occur when this agent is given in
combination with CYP-3A4 substrates with a narrow therapeutic index. One study
documented the occurrence of nephrotoxicity and neurotoxicity when nefazodone
was associated with the immunosuppressants cyclosporin and tacrolimus, and of
myositis and rhabdomyolysis with simvastatin.15,56 Concomitant use
of nefazodone and certain CYP-3A4 substrates, including cisapride, astemizole,
terfenadine, and loratadine, is contraindicated, as it may predispose patients
to torsades de pointes, a potentially fatal ventricular dysrhythmia
associated with marked electrocardiographic QTc prolongation.57
The Pharmacist's Role
There is no
comprehensive guide, chart, or computer software program to help clinicians
clearly and quickly identify or predict which drugs interact with CYP enzymes
and cause clinically significant drug interactions. More research and clinical
drug trials on these enzymes and their interactions need to be conducted and
reported. With this in mind, one way to help manage these drug interactions is
to have a basic understanding of the physiologic role CYP-450 enzymes play in
metabolizing drugs. With knowledge of how these enzymes work and what their
role is in drug interactions, pharmacists can better predict significant
interactions that are likely to occur and identify potential problematic
drugs.
An understanding of which
CYP-450 isoenzyme is responsible for the metabolism of a drug is essential for
trying to predict and understand the magnitude of drug interactions. Some
drug-metabolism inhibitors are highly selective for certain CYP isoenzymes.
Drugs that are highly selective enzyme inhibitors may also be substrates for
that same enzyme system and may cause an interaction by being a competitive
inhibitor. Obviously, if it is known that a new drug is metabolized by a
specific CYP isoenzyme system, it is logical to assume that the new drug will
exhibit drug interactions with known inducers and inhibitors of specific CYP
isoenzymes. Management of patients in a clinical setting may be simplified if
drugs that are known to produce harmful drug interactions with each other are
avoided or at least limited and the patient is closely monitored.
REFERENCES
1. Michalets EL.
Review of therapeutics. Update: clinically significant cytochrome P-450 drug
interactions. Pharmacotherapy. 1998;18:84-112.
2. Streetman DS.
Cytochrome P 450 enzymes. Alexandria, VA: National Community Pharmacists
Association; 1999.
3. Lamb E. Top 200
prescription drugs of 2006. Pharmacy Times. 2007;73:34, 36.
4. Sproule BA, Naranjo
CA, Bremner KE, et al. Selective serotonin reuptake inhibitors and CNS drug
interactions: a critical review of the evidence. Clin Pharmacokinet.
1997;33:454-471.
5. Guengerich FP. Role
of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol.
1997;43:7-35.
6. Nelson DR, Koymans
L, Kamataki T, et al. P450 superfamily: update on new sequences, gene mapping,
accession numbers and nomenclature. Pharmacogenetics. 1996;6:1-42.
7. Preskorn SH.
Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo,
OK: Professional Communications, Inc; 1996.
8. von Moltke LL,
Greenblatt DJ, Court MH, et al. Inhibition of alprazolam and desipramine
hydroxylation in vitro by paroxetine and fluvoxamine: comparison with other
selective serotonin reuptake inhibitory antidepressants. J Clin Psychopharm
. 1995;15:125-131.
9. Brown CH. Overview
of drug interactions modulated by cytochrome P450. US Pharmacist.
2001;4:HS26-45.
10. Ketter TA, Post RM,
Worthington K. Principles of clinically important drug interactions with
carbamazepine. Parts 1 and II. J Clin Psychopharmacol. 1991;11:198-203,
306-313.
11. Pfandl B, M^rike K,
Winne D, et al. Stereoselective inhibition of nortriptyline hydroxylation in
man by quinidine. Xenobiotica. 1992;22:721-730.
12. Lin JH, Lu AY.
Inhibition and induction of cytochrome P450 and the clinical implications.
Clin Pharmacokinet.1998;35:361-390.
13. Nemeroff CB, DeVane
CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J
Psychiatry. 1996;153:311-320.
14. Hamelin BA, Turgeon
J, VallÈe F, et al. The disposition of fluoxetine but not sertraline is
altered in poor metabolizers of debrisoquin. Clin Pharmacol Ther.
1996;60:512-521.
15. Spina E, Scordo MG,
D'Arrigo C. Metabolic drug interactions with new psychotropic agents.
Fundam Clin Pharmacol. 2003;17:517-538.
16. Bergstrom RF,
Peyton AL, Lemberger L. Quantification and mechanism of the fluoxetine and
tricyclic antidepressant interaction. Clin Pharmacol Ther.
1992;51:239-248.
17. Maes M, Westenberg
H, Vandoolaeghe E, et al. Effects of trazodone and fluoxetine in the treatment
of major depression. J Clin Psychopharmacol. 1997;17:358-364.
18. Avenoso A, Spina E,
Campo G, et al. Interaction between fluoxetine and haloperidol:
pharmacokinetic and clinical implications. Pharmacol Res.
1997;35:335-339.
19. Spina E, Avenoso A,
Facciolá G, et al. Effect of fluoxetine on the plasma concentrations of
clozapine and its major metabolites in patients with schizophrenia. Int
Clin Psychopharmacol. 1998;13:141-145.
20. Bondolfi G, Eap CB,
Bertschy G, et al. The effect of fluoxetine on the pharmacokinetics and safety
of risperidone in psychiatric patients. Pharmacopsychiatry.
2002;35:50-56.
21. Lemberger L, Rowe
H, Bosomworth JC, et al. The effect of fluoxetine on the pharmaco!= kinetics
and psychomotor responses of diazepam. Clin Pharmacol Ther.
1988;43:412-419.
22. Greenblatt DJ,
Preskorn SH, Cotreau MM, et al. Fluoxetine impairs clearance of alprazolam but
not of clonazepam. Clin Pharmacol Ther. 1992;52:479-486.
23. Jalil P. Toxic
reaction following the combined administration of fluoxetine and phenytoin:
two case reports. J Neurol Neurosurg Psychiatry. 1992;55:412-413.
24. Shader RI,
Greenblatt DJ, von Moltke LL. Fluoxetine inhibition of phenytoin metabolism.
J Clin Psychopharmacol. 1994;14:375-376.
25. Duncan D, Sayal K,
McConnell H, et al. Antidepressant interactions with warfarin. Int Clin
Psychopharmacol. 1998;13:87-94.
26. Walley T,
Pirmohamed M, Proudlove C, et al. Interaction of metoprolol and fluoxetine.
Lancet. 1993;341:967-968.
27. Drake WM, Gordon
GD. Heart block in a patient on propranolol and fluoxetine. Lancet.
1994;343:425-426.
28. Sternbach H.
Fluoxetine-associated potentiation of calcium channel blockers. J Clin
Psychopharmacol. 1991;11:390-391.
29. Spigset O, Granberg
K, Hâgg S, et al. Relationship between fluvoxamine pharmacokinetics and
CYP2D6/CYP2CI9 phenotype polymorphisms. Eur J Clin Pharmacol.
1997;52:129-133.
30. Xu ZH, Xie HG, Zhou
HH. In vivo inhibition of CYP2C19 but not CYP2D6 by fluvoxamine. Br J Clin
Pharmacol. 1996;42:518-521.
31. Spina E, Pollicino
AM, Avenoso A, et al. Effect of fluvoxamine on the pharmacokinetics of
imipramine and desipramine in healthy subjects. Ther Drug Monit.
1993;15:243-246.
32. Anttila AK, Rasanen
L, Leinonen EV. Fluvoxamine augmentation increases serum mirtaza!= pine
concentration three- to fourfold. Ann Pharmacother. 2001;35:1221-1223.
33. Daniel DG, Randolph
C, Jaskiw G, et al. Coadministration of fluvoxamine increases serum
concentrations of haloperidol. J Clin Psychopharmacol. l994;14:340-343.
34. Hiemke C, Weigmann
H, Hârtter S, et al. Elevated serum levels of clozapine after addition of
fluvoxamine. J Clin Psychopharmacol. 1994;14:279-281.
35. Weigmann H, Gerek
S, Zeisig A, et al. Fluvoxamine but not sertraline inhibits the metabolism of
olanzapine: evidence from a therapeutic drug monitoring service. Ther Drug
Monit. 2001;23:410-413.
36. Fleishaker JC,
Hulst LK. A pharmacokinetic and pharmacodynamic evaluation of the combined
administration of alprazolam and fluvoxamine. Eur J Clin Pharmacol.
1994;46:35-39.
37. Perucca E, Gatti G,
Cipolla G, et al. Inhibition of diazepam metabolism by fluvoxamine: a
pharmacokinetic study in normal volunteers. Clin Pharmacol Ther.
1994;56:471-476.
38. DeVane CL,
Markowitz JS, Hardesty SJ, et al. Fluvoxamine-induced theophylline toxi!=
city. Am J Psychiatry. 1997;154:1317-1318.
39. Yap KB, Low ST.
Interaction of fluvoxamine with warfarin in an elderly woman. Singapore Med
J. 1999;40:480-482.
40. Bertschy G, Baumann
P, Eap CB, et al. Probable metabolic interaction between methadone and
fluvoxamine in addict patients. Ther Drug Monit. 1994;16:42-45.
41. Eap CB, Bertschy G,
Powell K, et al. Fluvoxamine and fluoxetine do not interact in the same way
with the metabolism of the enantiomers of methadone. J Clin Psychopharmacol
. 1997;17:113-117.
42. Crewe HK, Lennard
MS, Tucker GT, et al. The effect of selective serotonin re-uptake inhibitors
on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin
Pharmacol. 1992;34:262-265.
43. Bloomer JC, Woods
FR, Haddock RE, et al. The role of cytochrome P4502D6 in the metabolism of
human liver microsomes. Br J Clin Pharmacol. 1992;33:521-523.
44. Brosen K, Hansen
JG, Nielsen KK, et al. Inhibition of paroxetine of desipramine metabolism in
extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol.
1993;44:349-355.
45. Alderman J,
Preskorn SH, Greenblatt DJ, et al. Desipramine pharmacokinetics when co!=
administered with paroxetine or sertraline in extensive metabolizers. J
Clin Psychopharmacol. 1997;17:284-291.
46. Ozdemir V, Naranjo
CA, Herrmann N, et al. Paroxetine potentiates the central nervous system side
effects of perphenazine: contribution of cytochrome P4502D6 inhibition in
vivo. Clin Pharmacol Ther. 1997;62:334-347.
47. Spina E, Avenoso A,
Facciolá MG, et al. Plasma concentrations of risperidone and
9-hydroxyrisperidone during combined treatment with paroxetine. Ther Drug
Monit. 2001;23:223-227.
48. Kobayashi K,
Ishizuka T, Shimada N, et al. Sertraline N-demethylation is catalyzed by
multiple isoforms of human cytochrome P450 in vitro. Drug Metab Dispos.
1999;27:763-766.
49. Preskorn SH,
Alderman J, Chung M, et al. Pharmacokinetics of desipramine coadministered
with sertraline or fluoxetine. J Clin Psychopharmacol. 1994;14:90-98.
50. Kurz DL, Bergstrom
RF, Goldberg MJ, et al. The effect of sertraline on the pharmacokinetics of
desipramine and imipramine. Clin Pharmacol Ther. 1997;62:145-156.
51. Kobayashi K, Chiba
K, Yagi T, et al. Identification of cytochrome P450 isoforms involved in
citalopram N-demethylation by human liver microsomes. J Pharmacol Exp Ther.
1997;280:927-933.
52. Otton SV, Ball SE,
Cheung SW, et al. Venlafaxine oxidation in vitro is catalyzed by CYP2D6. Br
J Clin Pharmacol. 1996;41:149-156.
53. Albers LJ, Reist C,
Vu RL, et al. Effect of venlafaxine on imipramine metabolism. Psychiatry
Res.2000;20:235-243.
54. Dahl ML, Voortman
G, Alm C, et al. In vitro and in vivo studies on the disposition of
mirtazapine in humans. Clin Drug Invest. 1997;13:37-46.
55. Rotzinger S, Baker
GB. Human CYP3A4 and the metabolism of nefazodone and hydroxy!= nefazodone by
human liver microsomes and heterologously expressed enzymes. Neuropsycho!=
pharmacol. 2002;12:91-100.
56. Thompson M, Samuels
S. Rhabdomyolysis with simvastatin and nefazodone. Am J Psychiatry.
2002;159:1607.
57. Abernethy DR,
Barbey JT, Franc J, et al. Loratadine and terfenadine interaction: both
antihistamines are associated with Qtc prolongation. Clin Pharmacol Ther.
2001;69:96-103.
To comment on this article,
contact
editor@uspharmacist.com.



