US Pharm. 2008;33(8):HS-9-HS-16.
The
pulmonary vasculature is normally a low-pressure system with approximately
one-tenth the resistance to flow of the systemic vasculature.1
Pulmonary hypertension (PH), a life-threatening condition with a poor
prognosis if untreated, is characterized by elevated mean pulmonary arterial
pressure (mPAP), which can lead to right ventricular failure.2 By
expert consensus, PH is defined as an mPAP greater than 25 mmHg at rest or 30
mmHg with exercise, as measured by right-heart catheterization.2
Data from 194 patients with idiopathic pulmonary arterial hypertension (IPAH)
in the National Institutes of Health registry from 1981 to 1985--before the
advent of disease-modifying therapy--found one-, three-, and five-year survival
rates to be 68%, 48%, and 34%, respectively; median survival was 2.8 years.3
Pulmonary hypertension is a
rare disorder, and epidemiologic data come largely from patients with IPAH. In
this subset of PH, the incidence rate is estimated to be two to five
individuals per million per year in the general population. The female:male
ratio is 1.7:1, and the third and fourth decades of life are the most common
time IPAH is diagnosed.1
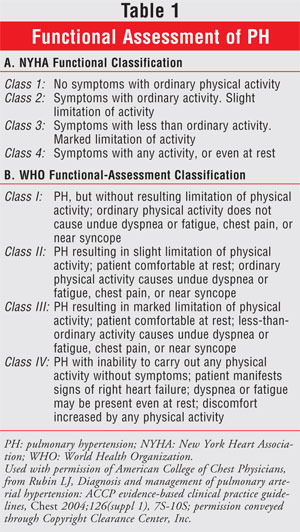
Most patients with PH
experience lethargy, fatigue, and exertional dyspnea. Measuring patients'
exercise capacity determines their World Health Organization (WHO) or New York
Heart Association (NYHA) functional status (TABLE 1).4
Eventually, right ventricular enlargement (cor pulmonale) and right-sided
heart failure will develop due to the increased pressure the heart must pump
against to send blood to the lungs, leading to symptoms such as angina,
syncope, peripheral edema, and abdominal distention. In addition, PH and
right-sided heart failure lead to characteristic changes in the heart sounds
detectable by auscultation.
Classification
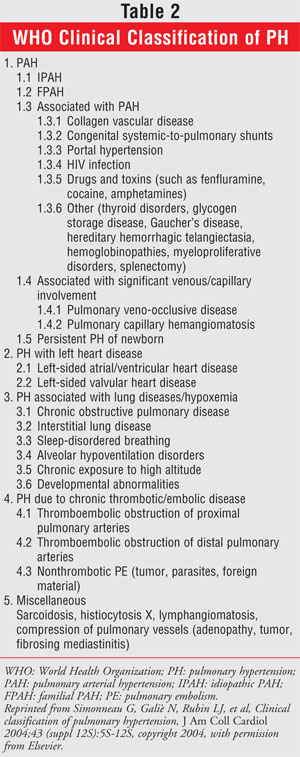
In recent years,
the classification scheme of PH has changed. Many clinicians continue to use
the terms IPAH (also known as primary PH) and secondary PH
(inconclusive of all cases of PH with known causes). The Second World
Symposium on Pulmonary Hypertension, held in 1998 in Evian, France,
recommended a new classification system; this system is now known as the Evian
classification and is used by the WHO. In 2003, this classification was
revised at the Third World Symposium (TABLE 2).5 The five
main PH categories are PAH, pulmonary venous hypertension, PH secondary to
respiratory-system disorders or hypoxemia, PH secondary to thrombotic or
embolic events, and a miscellaneous category consisting mainly of PH secondary
to diseases affecting the pulmonary vasculature.
Pathogenesis
The pathogenesis of PH is complex,
likely multifactorial, and imperfectly understood. However, agents now exist
that counteract the action (or supplement the lack thereof) of three molecular
pathways known to be implicated in PH--specifically, PAH. Prostacyclin synthase
in endothelial cells makes prostacyclin (PGI2); in pulmonary-artery smooth
muscle cells (SMCs), PGI2 stimulates adenylate cyclase to convert adenosine
triphosphate to the second messenger cyclic adenosine monophosphate (cAMP),
which is responsible for maintaining pulmonary-artery SMC relaxation and
inhibiting proliferation. These same endothelial cells also make nitric oxide
(NO), which induces guanylate cyclase to convert guanosine triphosphate to
cyclic guanosine monophosphate (cGMP), a second messenger with a mechanism of
action similar to that of cAMP. In PH, levels of cAMP and cGMP are diminished,
so administration of exogenous prostanoids (which increase cAMP levels) and
phosphodiesterase (PDE) inhibitors (which increase cGMP levels) are beneficial.
Endothelin (ET) A and B
receptors (ETA, ETB) also are known to be involved in
PAH, and the actions of ET-1 are mediated through them. Stimulation of ETA
receptors on pulmonary-artery SMCs results in sustained vasoconstriction and
cellular proliferation. In contrast, ETB receptors on endothelial
cells are theorized to be involved in the clearance of ET-1 in the vascular
beds. ETB receptor activation by ET-1 also leads to the release of
vasodilatory NO and PGI2, thereby offsetting the effects of the ETA
receptor. Selective and nonselective ET antagonists are available to
counteract these actions.
Additional causes of PH
include receptor and transporter genetic abnormalities; serotonergic
mechanisms including upregulation of the 5-hydroxytryptamine1B
receptor; downregulation of inhibition of voltage-dependent potassium channels
by hypoxia and agents such as fenfluramine; inflammation; procoagulant states;
and endothelial-cell dysfunction.1
Treatment
Treatment of PH
with disease-modifying therapy has been shown to improve hemodynamic measures,
WHO or NYHA functional class, and six-minute walking-test distance. Improved
survival has been found in uncontrolled trials and in a meta-analysis not
reaching statistical significance.6-9,10 Generally, however, loop
diuretics are considered for all patients with PH who have peripheral edema
and hepatic congestion. Oxygen therapy is administered to all WHO Class III
patients who have PH secondary to respiratory-system disorders. WHO Class I
patients also commonly receive oxygen, as do those with other classes, if
hypoxic. Warfarin is prescribed for all WHO Class IV (PH secondary to
thromboembolic disease) and typically Class I patients with IPAH and familial
PAH, given a recent review of seven observational trials (n = 488) showing a
mortality benefit.11 The goal international normalized ratio (INR)
is 2.0 (range 1.5–2.5), except for WHO Class IV patients, whose goal INR is
2.5 (range 2.0–3.0). Exercise training also has been shown to improve the
six-minute walking distance and WHO functional class in as few as four months.12
Some experts prescribe digoxin in patients with PH and refractory right
ventricular dysfunction. Some experts also advocate its use in patients with
PH taking calcium channel blockers (CCBs) to decrease the negative inotropic
effects of these agents.13 Data regarding the use of digoxin in
patients with PH and right ventricular dysfunction are limited and conflicting.14,15
Digoxin has not been shown to provide long-term benefit or improve survival in
patients with PH and right ventricular dysfunction.
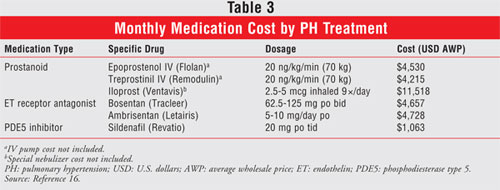
WHO Clinical Class I:
For PAH or WHO Class I, treatment focuses on using chronic pulmonary
vasodilator therapy (CCBs, prostanoids, ET receptor antagonists, and PDE type
5 [PDE5] inhibitors) to promote pulmonary SMC vasodilation and inhibit
cellular proliferation. It is not uncommon for patients to take one or a
combination of these therapies. The monthly cost for each drug therapy is
given in TABLE 3.16
Prior to initiating therapy,
all patients other than those with portopulmonary hypertension should undergo
right-heart catheterization for an acute vasodilator challenge to determine
whether they are candidates for CCB therapy. Vasodilatory agents used include
intravenous (IV) epoprostenol, IV adenosine, and inhaled NO. The test is
deemed positive if mPAP decreases by at least 10 mmHg and to less than 40
mmHg. Roughly 13% of patients have a positive response; of these, half will
have a sustained response with CCB therapy.17 CCBs should be
considered first-line therapy in patients with a positive acute vasodilator
response. Although trials suggest reduced mortality in PH with CCB use,
evidence suggesting their benefit is limited by the lack of randomized trials
comparing CCB therapy versus placebo in vasoreactive patients only.13,17
Trials with data supporting CCB use in PH used high-dose, sustained-release
therapy with titrations up to nifedipine 240 mg, amlodipine 20 mg, or
diltiazem 900 mg daily. Verapamil is generally avoided due to its negative
inotropic effects. Nonresponders to vasodilator testing or responders who
remain in NYHA functional Class III or IV should be considered for treatment
with prostanoids, ET receptor antagonists, or PDE5 inhibitors. The use of
these agents for PH other than PAH has been investigated in small,
uncontrolled, and often open-label studies and, as such, is often more art
than science for these indications.
Mechanistically, the
prostanoids overcome dysfunction in the endothelial SMCs' prostacyclin pathway
to increase cAMP levels. The synthetic prostacyclin epoprostenol (Flolan) is
indicated for the treatment of patients with NYHA functional Class III or IV
PAH and is considered first-line therapy in functional Class IV given the
small number of patients in trials of alternative agents. Compared with
historical control groups, epoprostenol increased absolute three-year survival
rates by approximately 20%.6,18 Epoprostenol is given via an IV
portable cassette infusion pump. It may be administered peripherally, but
central venous line administration is preferred to maintain IV line integrity.
Considering the drug's three- to-five-minute half-life, line loss or pump
malfunction can be a life-threatening emergency, and a second drug cassette
should always be prepared and a cassette pump be available to the patient. If
the backup cassette is not needed as a replacement, it should be used the
following day. The dose generally starts at 2 ng/kg/min and is titrated as
tolerated to as high as 200 ng/kg/min, although doses of 10 ng/kg/min to 50
ng/kg/min are more commonly seen. Adverse reactions include headache,
flushing, jaw pain, nausea, vomiting, diarrhea, hypotension, and anxiety.
Epoprostenol is supplied in 0.5-mg and 1.5-mg vials of powder for injection.
It must be diluted only with the special diluent. All of the prostanoid
products must be obtained through specialty pharmacy-distribution programs.
Treprostinil (Remodulin) is
another synthetic prostanoid used to treat PAH functional Class II–IV. It may
be administered by continuous subcutaneous infusion--which frequently causes
injection-site pain--or IV infusion. Treprostinil recently has been shown to
prolong survival in patients with PAH.19 Dosing and adverse
reactions are similar to those for epoprostenol. Treprostinil has two main
advantages over epoprostenol: (1) its four-hour half-life allows for
restarting the infusion with less urgency if the line is accidentally
discontinued, and (2) its commercially available solution is simpler to
prepare at home compared with the sterile manipulations required for the
powdered epoprostenol. Treprostinil solution for injection should be diluted
only with normal saline. Bacterial line infections, a serious consequence of
IV prostacyclin therapy, may occur more commonly with treprostinil.20
Iloprost (Ventavis) is an
inhaled prostacyclin that has been shown to improve NYHA functional class and
the six-minute walk test in doses of 2.5 mcg to 5 mcg nebulized six to nine
times daily.21 It has not been shown to increase survival in PH.
The FDA has approved iloprost for patients with PAH in NYHA functional Class
III–IV. Its adverse effects are similar to those of epoprostenol and also
include cough. Iloprost must be administered using a special nebulizer that
calibrates the specific dose.
The nonselective ET receptor
antagonist bosentan (Tracleer) and the selective ETA receptor
antagonist ambrisentan (Letairis) are oral agents used to treat PAH.
Sitaxsentan (Thelin), another selective ETA receptor antagonist, is
currently undergoing FDA review. Bosentan 62.5 mg to 125 mg twice daily and
ambrisentan 5 mg to 10 mg daily are indicated to improve exercise tolerance
and delay clinical progression in patients with WHO functional Class III–IV
and Class II–III symptoms, respectively. Bosentan also appears to have a
mortality benefit compared with historical controls.22 The main
adverse effect of ET receptor-antagonist therapy is hepatotoxicity,
necessitating frequent monitoring with liver-function tests. These agents are
teratogenic; women of childbearing potential should use two forms of
contraception while taking them. Ambrisentan, a CYP450 substrate at 3A4 and
2C19, has fewer drug interactions than bosentan, a substrate and inhibitor of
3A4 and 2C9. This difference is noteworthy in that bosentan decreases the
efficacy of both oral contraceptives and warfarin, whereas ambrisentan is not
known to do so. Both of these ET receptor antagonists require physician,
patient, and pharmacy enrollment in specialty pharmacy direct-shipment
programs to ensure ongoing monitoring for hepatotoxicity and pregnancy.
The PDE5 inhibitor sildenafil
(Revatio) inhibits metabolism of cGMP, resulting in relaxation and
antiproliferation of pulmonary endothelial SMCs, and is FDA-approved to
improve exercise tolerance in patients with PAH. Mortality rates with
sildenafil have not been assessed. Doses of 20 mg and up to 80 mg three times
daily have been tested in clinical trials.23 Adverse reactions
include headache, flushing, epistaxis, and dyspepsia; as in erectile
dysfunction, sildenafil for the treatment of PAH is contraindicated with
nitrates due to the possibility of severe hypotension. Interestingly, the PDE5
inhibitors tadalafil and vardenafil did not improve arterial oxygenation as
sildenafil did in patients with PAH and NYHA functional Class II–III during
short-term right heart catheterization.24
WHO Clinical Class II: Treatment
of PH due to left-sided cardiac disease (WHO Class II) centers on correcting
the underlying valvular, atrial, or ventricular abnormality. Chronic pulmonary
vasodilator therapy may be beneficial in select patients, such as those who
have undergone mitral valve replacement but still have PH postprocedurally.
Caution should be used, however; one trial of patients with severe left
ventricular dysfunction receiving standard therapy (n = 471) noted a trend
toward increased mortality in the epoprostenol group versus placebo.
Potentially, the positive inotropic effect of epoprostenol is detrimental to
an already-stressed heart, or the heart cannot compensate for the increased
flow across a newly dilated pulmonary vascular bed.25,26 Bosentan
also has been evaluated in PH that is due to systolic dysfunction and has not
been found to be beneficial.27,28
WHO Clinical Class III: In
WHO Class III PH, therapy consists of treating the underlying hypoxemia and
correcting it with chronic oxygen administration. For patients with PH and
obstructive sleep apnea, continuous positive airway pressure should be used.
Patients with PH secondary to living at high altitude should relocate to sea
level. Two trials in patients with chronic obstructive pulmonary disease and
partial pressure of oxygen (PaO2) between 55 mmHg and 60 mmHg found
that oxygen therapy decreases three- and five-year mortality.29,30
If these treatments are not completely successful and a patient remains in WHO
functional Class III–IV, chronic pulmonary vasodilator therapy may be
utilized. Small studies have shown benefit with sildenafil, NO, and iloprost,
whereas epoprostenol may increase hypoxemia.31-33
WHO Clinical Class IV: Up
to 3% of survivors of acute pulmonary embolism develop WHO Class IV PH.34
Anticoagulation to a goal INR of 2.0 to 3.0 is the primary therapy for these
patients. Thromboendarterectomy is an option for patients who are still
symptomatic despite anticoagulation. In addition, chronic pulmonary
vasodilator therapy may be used if these treatment modalities are suboptimal,
or as a bridge to surgery.
WHO Clinical Class V: The
treatment of WHO Class V PH is directed at the underlying etiology whenever
possible. Small studies have reported the use of prostanoids and ET receptor
antagonists.35,36
Conclusion
The treatment of PH
continues to evolve as additional drug-therapy targets are found. The severity
of the PH should always be determined prior to initiating treatment so that
patients' responses can be compared with baseline values. Primary therapy
(structural cardiac repair, chronic oxygen, anticoagulation, etc.) should be
directed at the underlying cause of the PH. If this is not possible, as in
PAH, or if the patient remains in WHO or NYHA functional Class II–IV,
vasoreactivity testing should be performed and a CCB prescribed if
appropriate. For patients with negative vasodilator tests or who become
nonresponsive to CCB therapy, prostanoids, ET antagonists, or PDE5 inhibitors
should be considered. Rarely, lung or heart–lung transplantation also has been
performed successfully in a curative manner.
REFERENCES
1. McLaughlin VV,
McGoon MD. Pulmonary arterial hypertension. Circulation.
2006;114:1417-1431.
2. Barst RJ, McGoon M,
Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial
hypertension. J Am Coll Cardiol. 2004;43(suppl 12S):40S-47S.
3. Rich S, Dantzker DR,
Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann
Intern Med.1987;107:216-223.
4. Rubin LJ. Diagnosis
and management of pulmonary arterial hypertension: ACCP evidence-based
clinical practice guidelines. Chest. 2004;126(suppl 1):7S-10S.
5. Simonneau G, Galiè
N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am
Coll Cardiol. 2004;43(suppl 12S):5S-12S.
6. Barst RJ, Rubin LJ,
McGoon MD, et al. Survival in primary pulmonary hypertension with long-term
continuous intravenous prostacyclin. Ann Intern Med. 1994;121:409-415.
7. McLaughlin VV,
Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact
of epoprostenol therapy. Circulation. 2002;106:1477-1482.
8. Sitbon O, Humbert M,
Nunes H, et al. Long-term intravenous epoprostenol infusion in primary
pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol.
2002;40:780-788.
9. Kuhn KP, Byrne DW,
Arbogast PG, et al. Outcome in 91 consecutive patients with pulmonary arterial
hypertension receiving epoprostenol. Am J Respir Crit Care Med.
2003;167:580-586.
10. Macchia A,
Marchioli R, Marfisi R, et al. A meta-analysis of trials of pulmonary
hypertension: a clinical condition looking for drugs and research methodology.
Am Heart J. 2007;153:1037-1047.
11. Johnson SR, Mehta
S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a
qualitative systematic review. Eur Respir J. 2006;28:999-1004.
12. Mereles D, Ehlken
N, Kreuscher S, et al. Exercise and respiratory training improve exercise
capacity and quality of life in patients with severe chronic pulmonary
hypertension. Circulation. 2006;114:1482-1489.
13. Rich S, Kaufmann E,
Levy PS. The effect of high doses of calcium-
channel blockers on
survival in primary pulmonary hypertension. N Engl J Med.1992;327:76-81.
14. Aubier M, Murciano
D, Viirès N, et al. Effects of digoxin on diaphragmatic strength generation in
patients with chronic obstructive pulmonary disease during acute respiratory
failure. Am Rev Respir Dis. 1987;135:544-548.
15. Rich S, Seidlitz M,
Dodin E, et al. The short-term effects of digoxin in patients with right
ventricular dysfunction from pulmonary hypertension. Chest.
1998;114:787-792.
16. Fleming T, ed. Red
Book 2007: Pharmacy's Fundamental Reference. Montvale, NJ: Thomson
Healthcare; 2007.
17. Sitbon O, Humbert
M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic
pulmonary arterial hypertension. Circulation. 2005;111:3105-3111.
18. Shapiro SM, Oudiz
RJ, Cao T, et al. Primary pulmonary hypertension: improved long-term effects
and survival with continuous intravenous epoprostenol infusion. J Am Coll
Cardiol. 1997;30:343-349.
19. Barst RJ, Galiè N,
Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients
treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195-1203.
20. Bloodstream
infections among patients treated with intravenous epoprostenol or intravenous
treprostinil for pulmonary arterial hypertension--seven sites, United States,
2003–2006. MMWR. 2007;56:170-172.
21. Olschewski H,
Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary
hypertension. N Engl J Med. 2002;347:322-329.
22. McLaughlin VV,
Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients
with primary pulmonary hypertension. Eur Respir J. 2005;25:244-249.
23. Galiè N, Ghofrani
HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial
hypertension. N Engl J Med.2005;353:2148-2157.
24. Ghofrani HA,
Voswinckel R, Reichenberger F, et al. Differences in hemodynamic and
oxygenation responses to three different phosphodiesterase-5 inhibitors in
patients with pulmonary arterial hypertension: a randomized prospective study. J
Am Coll Cardiol. 2004;44:1488-1496.
25. Califf RM, Adams
KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy
for severe congestive heart failure: the Flolan International Randomized
Survival Trial (FIRST). Am Heart J. 1997;134:44-54.
26. Montalescot G,
Drobinski G, Meurin P, et al. Effects of prostacyclin on the pulmonary
vascular tone and cardiac contractility of patients with pulmonary
hypertension secondary to end-stage heart failure. Am J Cardiol.
1998;82:749-755.
27. Packer M, McMurray
J, Massie BM, et al. Clinical effects of endothelin receptor antagonism with
bosentan in patients with severe chronic heart failure: results of a pilot
study. J Card Fail. 2005;11:12-20.
28. Kaluski E, Cotter
G, Leitman M, et al. Clinical and hemodynamic effects of bosentan dose
optimization in symptomatic heart failure patients with severe systolic
dysfunction, associated with secondary pulmonary hypertension--a multi-center
randomized study. Cardiology. 2007;109:273-280.
29. Nocturnal Oxygen
Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic
chronic obstructive lung disease: a clinical trial. Ann Intern Med.
1980;93:391-398.
30. Medical Research
Council Working Party. Long term domiciliary oxygen therapy in chronic hypoxic
cor pulmonale complicating chronic bronchitis and emphysema. Lancet.
1981;1:681-686.
31. Ghofrani HA,
Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and
pulmonary hypertension: a randomised controlled trial. Lancet.
2002;360:895-900.
32. Olschewski H,
Ghofrani HA, Walmrath D, et al. Inhaled prostacyclin and iloprost in severe
pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care
Med. 1999;160:600-607.
33. Strange C, Bolster
M, Mazur J, et al. Hemodynamic effects of epo prostenol in patients with
systemic sclerosis and pulmonary hypertension. Chest.
2000;118:1077-1082.
34. Pengo V, Lensing
AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary
hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257-2264.
35. Fisher KA, Serlin
DM, Wilson KC, et al. Sarcoidosis-associated pulmonary hypertension: outcome
with long-term epoprostenol treatment. Chest. 2006;130:1481-1488.
36. Steiner MK, Preston
IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion
therapy in pulmonary arterial hypertension: a pilot study. Chest.
2006;130:1471-1480.
To comment on this article, contact
rdavidson@jobson.com.



