USPharm. 2018;43(11):26-28.
ABSTRACT: Diabetic ketoacidosis (DKA) is a medical emergency caused by insulin deficiency. It is characterized by hyperglycemia, metabolic acidosis, and ketoacidosis. DKA arises from lack of insulin, with or without a precipitating event that leads to a cascade of pathophysiological changes. The goals of DKA treatment are to normalize volume status, hyperglycemia, electrolytes, and ketoacidosis. Pharmacists in community or ambulatory-care settings can assist in preventing DKA, while inpatient pharmacists play a role in management of DKA.
Diabetic ketoacidosis (DKA) is a serious medical emergency caused by insulin deficiency that takes a significant toll on the U.S. healthcare system.1,2 There are over 500,000 hospital days per year and $2.4 billion in medical costs attributed to DKA alone. DKA has high rates of morbidity and mortality, especially in younger type 1 diabetic patients. It is the most common cause of death for those under the age of 24 years with type 1 diabetes.3 It is estimated that 27% to 37% of patients with DKA are newly diagnosed with diabetes, usually type 1.1 Trauma, infection, or surgery may increase the risk of DKA in patients with type 2 diabetes.3 Mortality with DKA is generally associated with the underlying illness or comorbidity.1,3,4
Generally, DKA may be characterized by significant hyperglycemia, metabolic acidosis, and ketoacidosis. However, DKA may present in various ways, from euglycemia to severe hyperglycemia with or without dehydration and coma.3-5 The treatment approach for each patient is highly individualized based on a patient’s clinical factors.5
PATHOPHYSIOLOGY
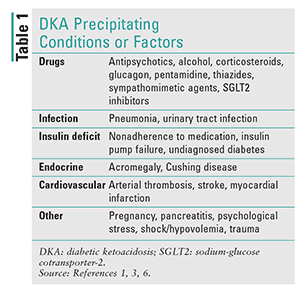
Simply put, DKA is caused by too little insulin and a response from the endocrine system that causes an increase in catecholamines, cortisol, glucagon, and growth hormone.4 The lack of insulin or other precipitating conditions (Table 1) stimulates the release of the regulatory hormones leading to hyperglycemia. The lack of glucose utilization and increase in gluconeogenesis and glycogenolysis causes hyperglycemia. Hyperglycemia further causes diuresis, leading to dehydration, electrolyte abnormalities, and kidney dysfunction.3,4 Because the body is unable to use glucose, lipase breaks down adipose tissue for energy, some of which is broken down into ketones, thereby leading to ketoacidosis.1,3
DIAGNOSIS
The typical signs and symptoms of DKA include polyuria, polydipsia, weight loss, nausea, vomiting, weakness, lethargy, altered mental status, acetone breath, tachycardia, hypotension, and deep hyperventilation.3,6 Some patients may present with rapid onset of DKA with no prior symptoms or hyperglycemia.
Laboratory parameters are crucial in the diagnosis of DKA. The initial workup should include plasma glucose, blood urea nitrogen, serum creatinine, electrolytes with anion gap, osmolality, serum ketones, arterial blood gas, CBC with differentials, urine ketones, urinalysis, chest x-ray, ECG, and urine, blood, and sputum cultures.3
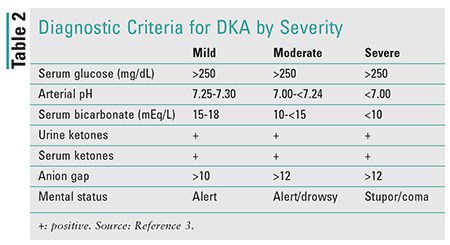
The 2009 American Diabetes Association consensus guideline outlines diagnostic criteria for DKA, classified according to severity (Table 2).3
TREATMENT
The goals of DKA treatment are to normalize fluid-volume status, hyperglycemia, electrolytes, and ketoacidosis. Any other potential precipitating factors should also be identified and addressed.
Fluid Replacement
In DKA, fluid loss can range from 6 to 9 L. Approximately one-half of the total volume loss should be replaced during the first 8 to 12 hours and the remaining volume within 24 to 36 hours. Rehydration is essential for tissue perfusion and resolution of the associated metabolic abnormalities. In addition, rehydration optimizes low-dose insulin therapy. Crystalloid fluids are preferred, with 1 to 1.5 L of 0.9% sodium chloride administered over the initial hour (15-20 mL/kg/hour).3,7 Caputo and colleagues conducted a prospective, randomized study in 27 patients; 14 patients received 0.9% saline solution at a rate of 1,000 mL/hour and 13 patients at a rate of 500 mL/hour.8 Both groups had similar biochemical characteristics at admission. No significant differences were found in any of the metabolic abnormalities, complications, or mortality rates between the two groups.8 Fluids should be replaced as quickly as possible, which can in turn lead to more cost-effective, shorter administration times.
Afterward, the rate of hydration should be guided by hydration status, hemodynamic status, electrolytes, and urinary output. Normal saline (0.9%) may be continued at 250 to 500 mL/hour (4-14 mL/kg/hour) in hyponatremic patients; half normal saline (0.45%) at similar infusion rates is optimal for patients who are eunatremic or hypernatremic.7 Five percent dextrose should be added to repletion fluids when the plasma glucose falls below 200 mg/dL to prevent hypoglycemia, while continuing insulin therapy at a decreased rate until ketonemia is resolved.3 Patients who have fluid retention (i.e., heart failure, chronic kidney disease) should be hydrated cautiously. Urine output is particularly important in monitoring these patients.
Insulin Therapy
Volume resuscitation is essential prior to initiating insulin therapy because insulin may worsen dehydration. Prior to initiation of insulin therapy, potassium should be at least 3.3 mEq/L. Insulin promotes the peripheral tissues’ utilization of glucose by diminishing gluconeogenesis and glycogenolysis and suppressing ketogenesis.3 Recommended insulin regimens include an initial IV bolus of regular insulin at 0.1 Unit/kg followed by continuous insulin infusion (at 0.1 Unit/kg/hour) or regular insulin as a continuous infusion at a rate of 0.14 Unit/kg/hour. If blood glucose does not fall by 10% in the first hour of insulin infusion, an additional bolus (0.1 Unit/kg or 0.14 Unit/kg, respectively) may be given and the infusion then continued at the previous rate. Once plasma glucose reaches 200 to 250 mg/dL, the insulin rate may be decreased by half, or to a rate of 0.02 to 0.05 Unit/kg/hour. In addition, dextrose should be added to the maintenance IV fluids at this point to prevent potential hypoglycemia.3,7,9
IV is the preferred route of administration of insulin in patients with DKA. A randomized, prospective trial conducted by Fisher and colleagues compared the use of low-dose insulin therapy by IV, IM, and SC routes.10 Forty-five patients were randomized in a 1:1:1 design. IV insulin displayed a more rapid decline in hyperglycemia (P <.01) and ketonemia (P <.05) in the first 2 hours of treatment compared with the IM and SC routes. The three groups showed similar responses 8 hours after treatment.10 The SC route of insulin has also been further investigated for DKA treatment. Insulin aspart and insulin lispro SC every 1 to 2 hours are as safe and effective as continuous insulin infusion in an ICU setting. Dosing regimens consist of 0.2 Unit/kg initially, followed by 0.1 Unit/kg every 1 hour or 0.3 Units/kg initially, followed by 0.2 Unit/kg every 2 hours until blood glucose is less than 250 mg/dL. At this point, insulin should be decreased to 0.05 to 0.1 Unit/kg, respectively, every 1 to 2 hours until the DKA is resolved. The time to DKA resolution, rate of hypoglycemia, and the length of hospital stay were similar between the two groups.11,12 The efficacy and cost effectiveness of SC rapid-acting insulin analogues have also been demonstrated for the treatment of uncomplicated mild-to-moderate DKA.13-15
Once DKA has resolved, patients who are capable of eating may be started on long-acting insulin for basal insulin requirements and a premeal rapid-acting insulin to control plasma glucose. Insulin infusion should be continued for at least an hour after the SC insulin is given to ensure that plasma insulin levels are adequate.3
Electrolytes
Potassium is generally depleted in DKA and other hyperglycemic emergencies. Insulin causes the intracellular movement of potassium into muscle cells by binding to its receptor on skeletal muscle. About two-thirds of patients will develop hypokalemia in the course of treatment for DKA.16 Potassium repletion should commence once the serum potassium falls below 5.3 mEq/L if patients have normal renal function. Twenty to 30 mEq of potassium may be supplemented to each liter of fluids. Patients with severe hypokalemia may require more potassium during the first hour of insulin treatment. Potassium chloride may cause hyperchloremic acidosis; therefore, potassium supplementation may be partially administered as potassium phosphate. If significant hypokalemia is present (<3.5 mEq/L), insulin therapy should be delayed until the potassium normalizes. Patients with hypokalemia should be monitored for arrhythmias.3
The use of bicarbonate is controversial owing to the lack of prospective, randomized clinical trials. Morris and colleagues investigated the use of bicarbonate therapy in DKA in 21 adults.17 They found that bicarbonate therapy did not provide any advantages in the rate of increase in pH or serum bicarbonate level in the blood or cerebrospinal fluid or in the decline of glucose or ketonemia. Rates of resolution of DKA between the two groups were also similar.17 There is currently no evidence to support the use of bicarbonate in DKA in patients with pH less than 6.9. It is recommended that the decision about whether to use bicarbonate be made based on the clinical state of the patient. Patients who present with severe acidosis with a pH less than 6.9 may be given 100 mmol of sodium bicarbonate in 400 mL sterile water with 20 mEq of potassium chloride at a rate of 200 mL/hour for 2 hours until the venous pH is greater than 7.0. This may be repeated every 2 hours until the pH is greater than 7.0. Patients who have a bicarbonate less than 10 or partial pressure of carbon dioxide (PCO2) less than 12 may experience deterioration of their pH and may be treated with bicarbonate as well.3,7
Phosphate supplementation is currently not recommended in most patients with DKA. Patients with DKA typically have about a 1 mmol/kg decrease in phosphate. Phosphate repletion has not shown any additional benefit for clinical outcome and could precipitate hypocalcemia.18,19 Patients with potential complications of hypophosphatemia (i.e., cardiac, muscle weakness, or rhabdomyolysis) may benefit from phosphate supplementation. Twenty to thirty mEq/L of potassium phosphate may be added to replacement fluids.3
THE PHARMACIST’S ROLE
Pharmacist involvement in DKA is multifactorial, depending on the specific area of practice. Pharmacists in an inpatient setting are well positioned to aid in the acute management of DKA. Pharmacists in the community or ambulatory-care setting have an important role in the prevention of DKA because they may be able to identify patients at high risk for DKA based on reasons such as medication nonadherence or insulin pump failures. Community pharmacists can also aid the patient in acquiring insulin when navigating prescription- plan coverage, or a lack of coverage, makes it difficult for the patient. Patient education is crucial regardless of practice setting. Every pharmacist can provide comprehensive medication education to DKA patients, stressing the importance of adherence to their medication regimen, addressing any barriers that may arise, and instructing on how to properly monitor blood glucose levels.
REFERENCES
1. Westerberg DP. Diabetic ketoacidosis: evaluation and treatment. Am Fam Physician. 2013;87(5):337-346.
2. Dhatariya KK, Vellanki P. Treatment of diabetic ketoacidosis (DKA)/hyperglycemic hyperosmolar state (HHS): novel advances in the management of hyperglycemic crises (UK versus USA). Curr Diab Rep. 2017;17(5):33.
3. Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343.
4. Tran TTT, Pease A, Wood A, et al. Review of evidence for adult diabetic ketoacidosis management protocols. Front Endocrinol (Lausanne). 2017;8:106.
5. American Diabetes Association. Diabetes care in the hospital: standards of medical care in diabetes—2018. Diabetes Care. 2018:41 (supp 1): S144-S151.
6. Misra S, Oliver NS. Diabetic ketoacidosis in adults. BMJ. 2015;351:h5660.
7. Nyenwe EA, Kitabchi AE. Evidence-based management of hyperglycemic emergencies in diabetes mellitus. Diabetes Res Clin Pract. 2011;94(3):340-351.
8. Caputo DG, Villarejo F, Valle GB, et al. Hydration in diabetic ketoacidosis. What is the effect of the infusion rate? Medicina (B Aires). 1997;57(1):15-20.
9. Gosmanov AR, Gosmanova EO, Dillard-Cannon E. Management of adult diabetic ketoacidosis. Diabetes Metab Syndr Obes. 2014;7:255-264.
10. Fisher JN, Shahshahani MN, Kitabchi AE. Diabetic ketoacidosis: low-dose insulin therapy by various routes. N Engl J Med. 1977;297(5):238-241.
11. Umpierrez GE, Cuervo R, Karabell A, et al. Treatment of diabetic ketoacidosis with subcutaneous insulin aspart. Diabetes Care. 2004;27(8):1873-1878.
12. Umpierrez GE, Latif K, Stoever J, et al. Efficacy of subcutaneous insulin lispro versus continuous intravenous regular insulin for the treatment of patients with diabetic ketoacidosis. Am J Med. 2004;117(5):291-296.
13. Della Manna T, Steinmetz L, Campos PR, et al. Subcutaneous use of a fast-acting insulin analog: an alternative treatment for pediatric patients with diabetic ketoacidosis. Diabetes Care. 2005;28:1856-1861.
14. Umpierrez GE, Jones S, Smiley D, et al. Insulin analogs versus human insulin in the treatment of patients with diabetic ketoacidosis: a randomized controlled trial. Diabetes Care. 2009;32:1164-1169.
15. Ersoz HO, Ukinc K, Kose M, et al. Subcutaneous lispro and intravenous regular insulin treatments are equally effective and safe for the treatment of mild and moderate diabetic ketoacidosis in adult patients. Int J Clin Pract. 2006;60:429-433.
16. Martin HE, Smith K, Wilson ML. The fluid and electrolyte therapy of severe diabetic acidosis and ketosis; a study of twenty-nine episodes (twenty-six patients). Am J Med. 1958;24:376-389.
17. Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986;105:836-840.
18. Winter RJ, Harris CJ, Phillips LS, et al. Diabetic ketoacidosis: induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67:897-900.
19. Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57:177-180.
To comment on this article, contact rdavidson@uspharmacist.com.


