US Pharm. 2023;48(1):HS11-HS16.
Drug-induced neurologic disorders may arise from unintended (iatrogenic) or unexpected effects on the central nervous system (CNS). Often, the mechanism of toxicity is an extension of the pharmacology of the drug. These disorders encompass a wide variety of conditions; those considered severe require admission to the ICU. Differentiating between symptoms and identification of the causative agent are essential for management. This article will focus discussion on serotonin syndrome (SS), neuromuscular malignant syndrome (NMS), malignant hyperthermia (MH), posterior reversible encephalopathy syndrome (PRES), and seizures—all of which are considered severe.
Serotonin Syndrome
SS is a severe, drug-induced toxidrome that causes central and peripheral intrasynaptic serotonin hyperactivity resulting in autonomic, behavioral, and neuromuscular symptoms (see TABLE 1).1-3 Enterochromaffin cells in the gut produce most of the body’s circulating serotonin, which stimulates vasoconstriction, gastrointestinal motility, and platelet aggregation.1 In the CNS, serotonin acts as an excitatory neurotransmitter that helps modulate affect, motor tone, aggression, and thermoregulation, among various other functions.1,3,4 The incidence is difficult to determine because of underreporting and misdiagnosis. A study found the incidence of SS was less than 0.1% in commercially insured patients using serotonergic agents in 2013, making it a rare occurrence.5

Initiation, increase, or addition of serotonergic agents increases the likelihood of developing SS. In a systematic review of 56 fatal SS cases, two concurrent serotonergic agents were the cause of 50% of patient deaths, followed by three or more concurrent serotonergic agents (34%).6 However, taking a single serotonergic agent accounted for 16% of the fatalities.6 Agents that inhibit serotonin reuptake include selective serotonin reuptake inhibitors (e.g., fluoxetine, citalopram), serotonin and norepinephrine reuptake inhibitors (e.g., venlafaxine, duloxetine), opioid analgesics (e.g., fentanyl, meperidine), serotonin modulators (e.g., trazodone, nefazodone), tricyclic antidepressants (e.g., amitriptyline, nortriptyline), tetracyclic antidepressants (e.g., amoxapine), 5-HT3 receptor antagonists (e.g., ondansetron, granisetron), St. John’s wort, cocaine, lysergic acid diethylamide (also known as LSD), and 3,4-methyl-enedioxy-methamphetamine (also known as MDMA).3,4,6,7 Agents that agonize serotonin receptors include antiepileptics (e.g., valproic acid, carbamazepine), buspirone, ergot alkaloid derivatives (e.g., methylergonovine, ergotamine), and lithium.3,4,6,7 Agents that increase serotonin release include amphetamines and amphetamine derivatives (e.g., cathinone, phentermine), dextromethorphan, dopamine agonists (e.g., ropinirole, rotigotine), ethanol, mirtazapine, and phenylethylamine.3,4,6,7
Assessment of autonomic instability, mental status changes, and neuromuscular hyperactivity allows for differential diagnosis of SS.1,3,4 Symptoms may range from mild (i.e., anxiety, diaphoresis, hyperreflexia, myoclonus, or tremor) to severe (i.e., coma, respiratory failure, rigidity, severe hyperthermia [³104°F/40°C], or tonic-clonic seizures).1,3,4 Due to the lack of confirmative diagnostic testing, the diagnosis is made via the exclusion of other disease states and the use of Hunter Serotonin Toxicity Criteria.1 These criteria include the use of a serotonergic agent along with observed/inducible clonus and/or agitation and hyperreflexia.1,3,4 Symptom onset can be within minutes to hours of ingesting a serotonergic agent in treatment-naive patients or after a dose increase.1,3,4
First-line treatment is the cessation of the causative agent, followed by supportive care targeting agitation, hyperthermia, and autonomic instability.1 Nonpharmacologic treatment options include cooling measures, oxygen, IV fluids, and continuous cardiac monitoring. In patients with severe agitation, benzodiazepines (e.g., an initial dose of diazepam 10 mg to 20 mg IV, repeated every 5-10 minutes until therapeutic sedation) should be considered.3,4,8 If severe symptoms continue, a serotonin (5HT-2A) receptor antagonist (e.g., an initial dose of cyproheptadine 12 mg oral, then 2 mg oral every 2 hours if symptoms persist [maximum daily dose of 32 mg]) may be utilized.3,4,8 If the patient has monoamine oxidase inhibitor–induced hypotension, direct-acting sympathomimetics are suggested.4 If the patient’s hypertension and tachycardia are severe, short-acting cardiovascular agents should be used.3,4,8 Nondepolarizing agents may be used in patients with severe hyperthermia, and IV intralipid therapy may be used in hemodynamic instability that is unresponsive to other treatments.3,4,8
There are several medications that should be avoided when treating a patient with SS. Antipyretics such as nonsteroidal anti-inflammatory drugs (NSAIDs) likely have no benefit because the hyperthermia stems from muscle contractions and spasms, not from an immunological response. Dantrolene has been proven ineffective in alleviating clonus and spasms.3,4,8 Propranolol potentially causes tachycardia and worsening hypotension.3,4,8 Bromocriptine might compound the serotoninergic activity of SS because it is also a serotonergic agent. Succinylcholine has a risk of inducing hyperkalemia and arrhythmias.3,4,8
Neuroleptic Malignant Syndrome
NMS is a severe adverse drug reaction commonly associated with the use antidopaminergic medications (see TABLE 2). It is commonly seen with the use of antipsychotics and has been associated with medications that antagonize dopamine-2 (D2) in the nigrostriatal, hypothalamic, and mesolimbic/cortical pathways or withdrawal of dopamine agonists, such as levodopa.5 The pathogenesis of NMS is unknown. Several proposed theories exist: excessive dopamine blockade at the D2 receptors, genetic polymorphism at the D2 receptor, sympathoadrenal hyperactivity, and predisposition of musculoskeletal fibers. The reported incidence of NMS is unknown but is estimated to be 0.03% with second-generation antipsychotics and associated with mortality of 5.6%.5

The average time to onset is within the first week after initiating a new medication.6 Common symptoms of NMS include “lead pipe” muscle rigidity, hyperthermia, altered mental status, and autonomic instability, with an elevated creatine kinase.5 Other latent symptoms may include dysphagia and sialorrhea (more prominent if caused by clozapine).5
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), criteria for NMS are hyperthermia (oral temperature >100.4˚F/38.0˚C on at least two occasions) and generalized muscle rigidity, or two or more of either elevated creatinine phosphokinase, changes in mental status, tachycardia, diaphoresis, blood pressure elevation or blood pressure fluctuation, urinary incontinence, pallor, or tachypnea.7 Conditions in which muscle rigidity and/or hyperthermia are prominent, such as CNS infections, lithium intoxication, heat shock, lethal catatonia, central anticholinergic syndrome, and malignant hyperthermia must be ruled out in the differential diagnosis.7
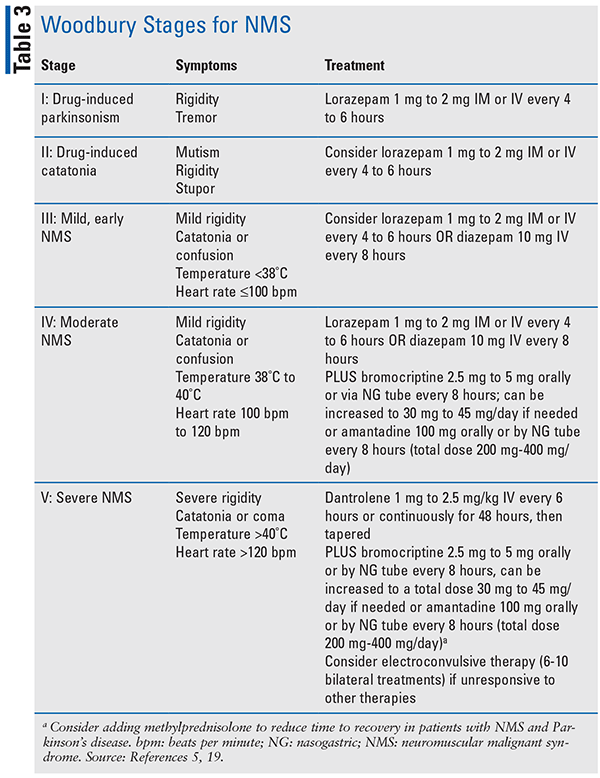
Treatment is based on severity which is classified by the Woodbury Stages (see TABLE 3). For all stages it is advised to provide supportive care and reduce or discontinue all suspected offending agents.5 For patients with stages IV–V, management includes the addition of IV fluids and cooling measures. Bromocriptine is used in stages IV and V and acts as an agonist at D2 receptors to reverse any excessive blockade of dopamine. Dantrolene is used for treatment of stage V for patients experiencing hyperthermia. It works by acting on skeletal muscle, causing relaxation by interference with the release of calcium ions.5
Malignant Hyperthermia
MH is a rare, drug-induced, genetic skeletal muscle condition, most often occurring in the operating room. It has been estimated to occur in between 1:10,000 and 1:150,000 general anesthetics. The highest proportion of cases occurs in men compared with women and in pediatric patients. Mortality rates for MH are approximately 4%.8
MH is a heritable trait and is closely linked with certain genetic variants. High-risk status can be confirmed if genetic testing is available and those genetic variants pathogenic for MH are identified.8 Triggering agents include all halogenated inhalational anesthetic agents (e.g., isoflurane, sevoflurane, desflurane) and the depolarizing neuromuscular blocker succinylcholine.9
The initial presentation results from increased metabolic activity within skeletal muscle cells as a compensation for the increased calcium released by the triggering agents. This leads to an increase in oxygen consumption and carbon dioxide production. The classic diagnostic features of MH include an unexplained, unexpected increase in end-tidal carbon dioxide, heart rate, and body temperature.8 Onset can occur as quickly as 10 minutes within exposure to triggering agents or be delayed for several hours. Generalized muscle rigidity is typically an indication the MH will likely be irreversible. Additional complications include respiratory acidosis, hyperkalemia, arrythmias, myoglobinuria, disseminated intravascular coagulation, and compartment syndrome.8
Management includes immediately reversing the reaction and treating the associated symptoms (see TABLE 4). Continue until metabolic stability for 24 hours, core temperature <100.4°F/38°C, creatine kinase continues to decrease, no evidence of ongoing myoglobinuria, and muscle rigidity has abated.10 Less evidence exists for antipyretics such as acetaminophen or NSAIDs, as hyperthermia is associated with excessive heat production by skeletal muscle so there is a lack of theoretical benefit.10 Dantrolene and hyperventilation should be the treatment priority and, if implemented rapidly, can prevent the development of life-threatening hyperthermia.

Posterior Reversible Encephalopathy Syndrome
PRES is a severe neurologic condition commonly seen in young or middle-aged adults, with a higher incidence in women.11 Mortality is estimated in 3% to 6% of cases if not properly treated. Most patients can make a full recovery.12
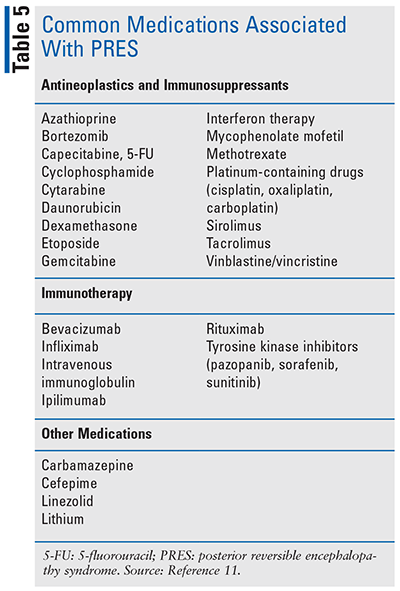
Common causes of PRES include hypertension, renal failure, eclampsia, sepsis, autoimmune disorders, and exposure to several medications (see TABLE 5).11 Development of PRES is thought to be due to a combination of disordered cerebrovascular autoregulation, blood-brain barrier breakdown leading to plasma extravasation, and release of cytokines that increase vascular permeability leading to interstitial vasogenic edema.12
Symptoms associated with PRES are encephalopathy, seizures, headaches, and visual symptoms.11 PRES is a clinical radiological diagnosis that must be made on presenting symptoms and risk factors and supported by imaging (CT and MRI). The hallmark finding on MRI is bilateral hyperdensity, typically in the parieto-occipital regions, indicating vasogenic edema.11
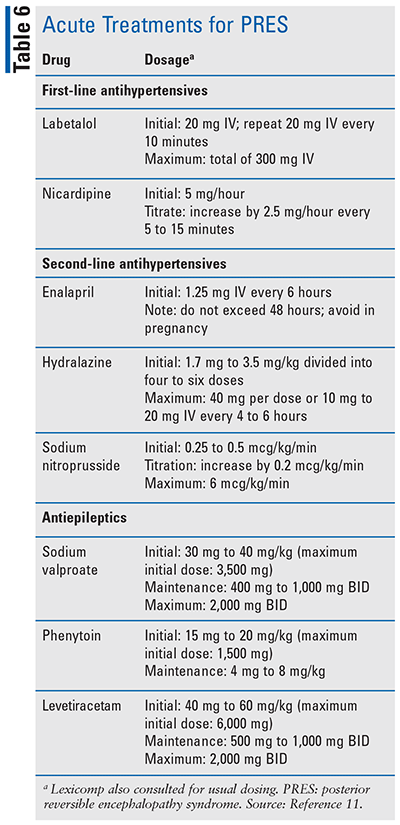
Management of PRES includes supportive care and removing or reversing the suspected cause. Supportive care includes correcting any electrolyte abnormalities, providing fluid resuscitation, and gradually reducing acute hypertension with a goal of 20% to 25% in the first few hours.11 Potential pharmacotherapies are listed in TABLE 6.
Seizures
Drug-induced seizures (DIS) may occur following acute or chronic therapy or abrupt withdrawal of many medications, including CNS agents, antibacterials, antivirals, cardiovascular agents, immunosuppressives, and NSAIDs, as well as toxins and poisons.13-16 Although DIS may be self-limiting, prolonged seizure activity may cause hypoxia, hypotension, pulmonary aspiration, hyperthermia, rhabdomyolysis, and metabolic acidosis.14 The incidence of DIS is estimated to be 6% of new-onset seizures and up to 9% of patients with status epilepticus.14
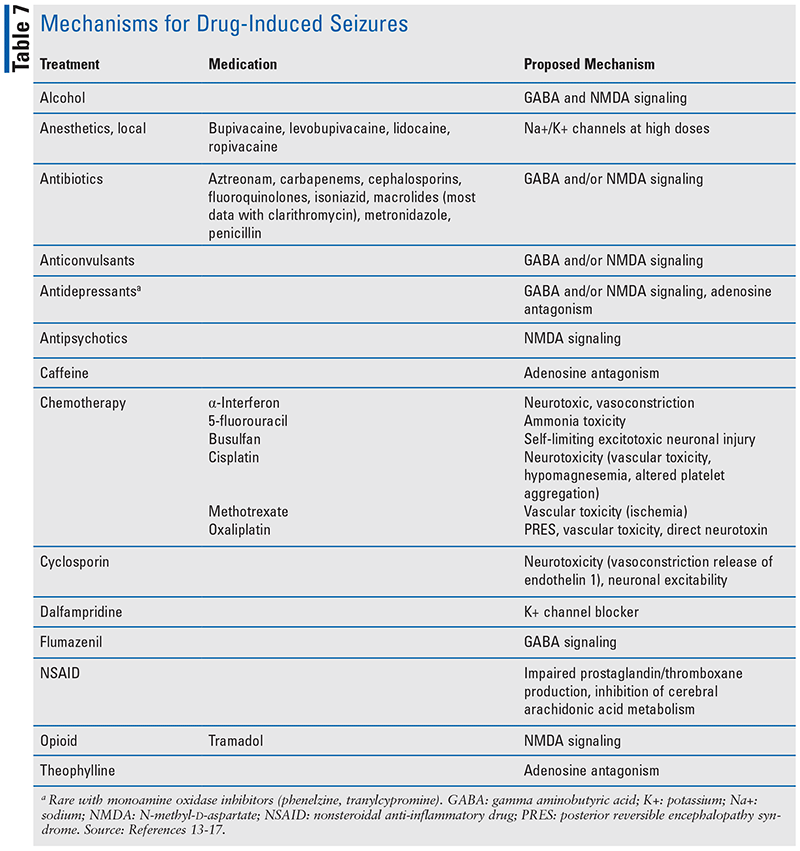
The mechanism by which drugs induce seizures varies (see TABLE 7). Altered electroencephalogram activity may result from abnormal neuronal discharges of excitatory (i.e., glutamate, N-methyl-d-aspartate) or inhibitory (i.e., gamma aminobutyric acid, glycine) neurotransmitters. Reducing inhibitory or decreasing excitatory pathways induces seizures.13,14 Other neurotransmitters known to play a role in seizures include norepinephrine, dopamine, serotonin, acetylcholine, histamine, and adenosine.14 In addition, seizures may result from abnormalities of blood perfusion and oxygenation to the CNS, as well as metabolic disturbances (e.g., hypomagnesemia, hyponatremia, hypoglycemia).14
Initial treatment involves stabilizing the patient’s blood pressure and heart rate, providing adequate oxygenation, and correcting any metabolic imbalances.14,17 Initial anticonvulsant therapy includes IV benzodiazepines. Lorazepam 4 mg every 4 to 5 minutes is preferred; however, midazolam 5 mg IM may be used if IV access is not available. For isoniazid-induced seizures, pyridoxine should be added, dosing gram-for-gram based on isoniazid amount or 25 mg/kg IV with a maximum of 5 g.
Conclusion
Neurotoxicity from drugs can be severe and, if not properly treated, lead to fatal outcomes. It is critical to be able to identify signs and symptoms associated with each different toxicity so that appropriate treatment may be implemented. In most drug-induced toxicities, the first approach will be removing the causative agent and providing supportive care. Pharmacists play an integral role in identifying drug-induced toxicities and knowing the various treatment strategies.
REFERENCES
1. Francescangeli J, Karamchandani K, Powell M, Bonavia A. The serotonin syndrome: from molecular mechanisms to clinical practice. Int J Mol Sci. 2019;20(9):2288.2. Prakash S, Rathore C, Rana K, Patel H. Antiepileptic drugs and serotonin syndrome—a systematic review of case series and case reports. Seizure. 2021;91:117-131.
3. Prakash S, Rathore C, Rana K, Prakash A. Fatal serotonin syndrome: a systematic review of 56 cases in the literature. Clin Toxicol (Phila). 2021;59(2):89-100.4. Wang RZ, Vashistha V, Kaur S, Houchens NW. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
5. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.6. Medication-induced movement disorders and other adverse effects of medication. In: Diagnostic and Statistical Manual of Mental Disorders. DSM Library. American Psychiatric Association Publishing; 2022. https://dsm-psychiatryonline-org.dml.regis.edu/doi/10.1176/appi.books.9780890425787.Medication_Induced_Movement_Disorders.
7. Tse L, Barr AM, Scarapicchia V, Vila-Rodriguez F. Neuroleptic malignant syndrome: a review from a clinically oriented perspective. Curr Neuropharmacol. 2015;13(3):395-406.8. Hopkins PM, Girard T, Dalay S, et al. Malignant hyperthermia 2020: guideline from the Association of Anaesthetists. Anaesthesia. 2021;76(5):655-664.
9. Kim KSM, Kriss RS, Tautz TJ. Malignant hyperthermia: a clinical review. Adv Anesth. 2019;37:35-51.10. Litman RS, Smith VI, Larach MG, et al. Consensus statement of the Malignant Hyperthermia Association of the United States on unresolved clinical questions concerning the management of patients with malignant hyperthermia. Anesth Analg. 2019;128(4):652-659.
11. Triplett JD, Kutlubaev MA, Kermode AG, Hardy T. Posterior reversible encephalopathy syndrome (PRES): diagnosis and management. Pract Neurol. 2022;22(3):183-189.12. Gewirtz AN, Gao V, Parauda SC, Robbins MS. Posterior reversible encephalopathy syndrome. Curr Pain Headache Rep. 2021;25(3):19.
13. Larson EA, Accardi MV, Zhong Y, et al. Drug-induced seizures: considerations for underlying molecular mechanisms. Int J Toxicol. 2021;40(5):403-412.14. Chen HY, Albertson TE, Olson KR. Treatment of drug-induced seizures. Br J Clin Pharmacol. 2016;81(3):412-419.
15. Wanleenuwat P, Suntharampillai N, Iwanowski P. Antibiotic-induced epileptic seizures: mechanisms of action and clinical considerations. Seizure. 2020;81:167-174.16. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82.
17. Skolnik A, Monas J. The crashing toxicology patient. Emerg Med Clin North Am. 2020;38(4):841-856.18. Oruch R, Pryme IF, Engelsen BA, Lund A. Neuroleptic malignant syndrome: an easily overlooked neurologic emergency. Neuropsychiatr Dis Treat. 2017;13:161-175.
19. Strawn JR, Keck PE Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164(6):870-876.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



