US Pharm. 2009;34(1)(Oncology suppl):7-9,14.
ABSTRACT: Sickle cell disease (SCD) is the most common autosomal, recessive, inherited blood disorder in the United States. SCD primarily affects African Americans, although it is also found in Hispanics and people of Middle-Eastern ancestry. Stroke in SCD can be overt or silent. The standard of care for pediatric stroke is chronic transfusion to decrease the level of hemoglobin S. However, treatment with chronic transfusion is not without complications. In recent years, ongoing studies have used hydroxyurea to decrease the complications of SCD and reduce organ failure in children. This drug has also played a role in decreasing overt and silent stroke.
In the United States, there are approximately 50,000 people with homozygous sickle cell disease (SCD), the most severe of the sickle cell disorders.1 Hemoglobin S is the main hemoglobin in SCD. It is different from normal hemoglobin A in that hemoglobin S, when deoxygenated, will turn into rodlike polymers that are very rigid. Sickle cell polymerization causes the red blood cell to become sickle-like and elongated. These sickle cells are sticky and rigid and have difficulty circulating in the bloodstream. This circulation difficulty leads to clumping of cells and obstruction of blood vessels, causing vaso-occlusion and producing pain and infarction of the tissue involved. The result is a painful episode or crisis.
Sickling can also affect the lungs, resulting in a condition called acute chest syndrome. Acute chest syndrome etiology is unknown, but is defined as a new pulmonary alveolar consolidation that involves at least one complete lung segment. The patient will have fever, chest pain, increased respiratory rate, wheezing, and/or coughing. Acute chest syndrome is the leading cause of death among patients with SCD.2
Bone damage from sickling may result in avascular necrosis. Leg ulcers occur in SCD from the inability to heal as well as from poor circulation of the lower extremities. When circulating blood becomes trapped in the spleen, causing massive expansion of the organ, it is known as splenic sequestration. As a result, the child’s hemoglobin and/or platelets decrease drastically, leading to a medical emergency. Additional organ damage from sickling in the spleen causes a functional asplenia in early childhood. Children who are not placed on prophylactic penicillin in infancy are at risk for infections with encapsulated organisms, which can result in sepsis and death. SCD affects the kidneys, causing hematuria, papillary necrosis, proteinuria, and glomerular injury.3 Eye injury may also occur from sickling and vaso-occlusion in the retinas of children with SCD.4
For children under 20 years of age with homozygous SCD, the prevalence of overt stroke is 7.8% to 11%, while silent stroke has a prevalence rate of 17%.5-8 Overt stroke shows obvious signs of neurologic deficits, whereas silent stroke shows MRI changes in the brain without physical findings.9
Standards of Care
The treatment of SCD has improved markedly over the last decades. Newborn screening for hemoglobinopathies has allowed the medical community to identify children with SCD in early infancy, before the child has his or her first vaso-occlusive episode. Most children identified by newborn screening are referred to major medical centers that have a Comprehensive Sickle Cell Disease Program. With early parent teaching on how to care for a child with SCD and the use of prophylactic penicillin, the rate of deaths due to sepsis before the age of 5 years has decreased markedly.10
Chronic Transfusion: Stroke in children with SCD is caused by impaired circulation due to narrowing or obstruction of the major cranial vessels and ischemia as a result of sickling. These children have significant physical impairments, as well as considerable neuropsychological and cognitive deficits.11 A child with an overt stroke should start on a chronic transfusion program to increase hemoglobin A and decrease the concentration of sickle hemoglobin, thereby reducing the risk of a second stroke. Children who are not treated with chronic transfusions have an 80% chance of a second stroke within three years of the previous one.12 Chronic transfusion decreases the risk of stroke but does not eliminate it.13 Transfusion has multiple complications, including iron overload, which can lead to organ damage of the heart, kidneys, and liver. To reduce iron overload, patients require a chelating agent such as subcutaneous deferoxamine (Desferal) or oral deferasirox (Exjade).
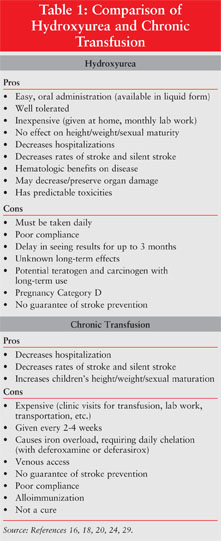
A main goal of care is to prevent stroke in these children by identifying those at risk. It is now documented that the increased velocity of the intracranial arteries is a precursor to overt stroke.14 A large multi-institutional study showed that chronic transfusion greatly reduces the risk of a first stroke in children with SCD who have abnormal results on transcranial Doppler (TCD) ultrasonography.15 Obtaining yearly TCD ultrasounds to evaluate the velocity of the cranial arteries is now a standard of care; those children with abnormal velocities should start on a chronic transfusion program as well. TCD ultrasonography is especially helpful with children since it is noninvasive, pain free, requires no sedation, and is easy to perform by trained personnel. The neurologic injuries from silent stroke have lead to the recognition of learning disabilities, memory problems, and lower IQs in these children, causing another major concern for the student, parent, teacher, and medical community.11 The benefits and drawbacks of chronic transfusion are described in Table 1.
Transplant: A bone marrow or stem cell transplant can cure SCD. Unfortunately, only 10% to 15% of children with SCD have human leukocyte antigen (HLA)-matched siblings who do not have SCD themselves.16 This procedure is highly invasive and prone to complications, such as graft-versus-host disease.
Hydroxyurea: Hydroxyurea is the only FDA-approved chemotherapeutic agent for the treatment of SCD in adults.17 Approval was based on the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. The double-blind, randomized, placebo-controlled study was closed early because of the beneficial effects noted in the patients taking hydroxyurea. These benefits included a 44% reduction in painful episodes, 58% reduction in hospitalizations for painful episodes, reduced chest syndrome events, and a decrease in the number of blood transfusions given to patients.18
Hydroxyurea has been used off label in children with SCD for many years. Many sickle cell centers use hydroxyurea in children with severe disease who have not had overt strokes but who do have several hospital admissions a year for painful episodes and chest syndrome, resulting in poor quality of life. As in adults, hydroxyurea raises hemoglobin F, which does not have problems with deoxygenation, and decreases the number of white blood cells— the reticulocyte and platelet counts that are usually increased in children with SCD. All these benefit the child the same as the adult, resulting in fewer hospitalizations and vaso-occlusive events, improving organ function, and lessening chest syndrome.19
Hydroxyurea is a chemotherapeutic agent used in treating malignancies, which has caused concerns about its carcinogenic and mutagenic effects. To date, a trial following children for seven years on hydroxyurea revealed that no patient in the study acquired myelodysplasia, leukemia, or any other malignancy.20 However, long-term adverse effects of hydroxyurea use in children require additional investigation. The use of hydroxyurea needs to be weighed cautiously (TABLE 1), and children should be monitored by a pediatric hematologist.
Hydroxyurea Therapy
Hydroxyurea has been used for over 40 years in the treatment of leukemia, melanoma, polycythemia vera, and ovarian cancer.21,22 Clinicians observed that the drug had the ability to increase fetal hemoglobin that does not polymerize with deoxygenation as does hemoglobin S. This led to several clinical trials in the 1980s that resulted in the FDA approval of hydroxyurea for use in adults in 1998.
Hydroxyurea acts on the sickle hemoglobin by inhibiting ribonucleotide reductase.1,17,18,23 When this process is blocked, production of sickle cells from rapidly dividing mature precursor cells cannot proceed, resulting in the destruction of the cell.1 The cell line is then forced to produce cells from primitive progenitor cells, which produce red blood cells with high contents of hemoglobin F at a slower pace. Hemoglobin F is further increased by the metabolism of hydroxyurea and the production of nitric oxide that in turn stimulates guanylate cyclase, which makes more fetal hemoglobin.1 Since hydroxyurea interferes with cell division, it has the potential of being teratogenic and carcinogenic.24 Another effect of hydroxyurea is myelosuppression.25
Hydroxyurea is rapidly absorbed with peak serum concentrations seen within 120 minutes.23,24 It distributes throughout the cells and enters all body compartments including the central nervous system, with approximately 50% metabolized by the liver. The metabolites are excreted as carbon dioxide through the lungs
and as urea by the kidneys. The remaining 50% is excreted as unchanged drug by the kidneys. The elimination half-life is 3.5 to 4.5 hours.23,24
Dosing of hydroxyurea therapy in children with SCD is started at 15 mg/kg/day once daily.1 Renal impairment requires dose reduction. Dosages are titrated to response and to avoid hematologic and renal toxicity. Hematologic parameters need to be reassessed after two weeks. This should result in a reduction of white blood cells and platelet counts. If they do not, the dose is reevaluated and recalculated. With monthly complete blood count monitoring, renal and hepatic function are also assessed for signs of adverse events such as elevated liver enzymes and increased blood urea nitrogen and creatinine levels.
Every 12 weeks, the dosage is reassessed to either increase by 5 mg/kg/day or to be held until the patient’s hematologic parameters return to within range. The maximum tolerated dose in children is 35 mg/kg/day.1 After about six months, most patients stabilize on their individualized dose.
Hydroxyurea is contraindicated in pregnancy (Pregnancy Category D), severe anemia, hypersensitivity to any component of the formulation, and severe bone marrow suppression.24 The drug is mutagenic and clastogenic, and long-term use could result in secondary leukemia. As hydroxyurea may increase the toxicity of didanosine, concomitant use is not recommended. It is available in tablet or capsule form. Capsules may be opened and emptied into water. The drug may also be compounded into a suspension for use in children.
Studies Related to Stroke and Hydroxyurea in Children
Since the early 1990s, major sickle cell centers around the country have pooled their data for the study of SCD. These centers formed the Cooperative Study of Sickle Cell Disease. The National Institute of Health’s National Heart, Lung, and Blood Institute sponsored many of these research studies. Further investigations are needed in order to prove that hydroxyurea can decrease TCD velocities in children with SCD.13,26
Two studies of infants with SCD (HUG-KIDS) have shown that hydroxyurea is safe for use in children, with no effects on growth, height, or sexual maturation, and is associated with only limited and manageable hematotoxicity, the same as in adults.27,28 The second of these studies is ongoing and continues to evaluate the efficacy of this drug in children and its toxicities. More research is needed and ongoing. A summary of hydroxyurea trials in children is provided in TABLE 2.

Conclusion
Children with SCD, specifically homozygous hemoglobin S disease, experience many complications, with stroke being one of the most devastating. Through research and better supportive care over the last few decades, these children can now enjoy a better quality of life. More recently, hydroxyurea has been added as an oral agent to decrease complications of SCD and preserve organ function in children. However, further research is warranted for pediatric use.
REFERENCES
1. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358:1362-1369.
2. Vichinsky EP, Neumeyer LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med. 2000;342:1865-1885.
3. Falk RK, Jennette JC. Renal disease. In: Embury S, Hebble R, Mohandas N, Steinberg M, eds. Sickle Cell Disease Basic Principles and Clinical Practice. Philadelphia, PA: Lippincott-Raven; 1996:673-680.
4. Lutty GA, Goldberg MF. Ophthalmologic complications. In: Embury S, Hebble R, Mohandas N, Steinberg M, eds. Sickle Cell Disease Basic Principles and Clinical Practice. Philadelphia, PA: Lippincott-Raven; 1996:703-724.
5. Balkaran B, Char G, Morris JS, et al. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr. 1992;120:360-366.
6. Pegelow CH, Macklin EA, Moser FG, et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. 2002;99:3014-3018.
7. Moser FG, Miller ST, Bello JA, et al. The spectrum of brain MR abnormalities in sickle-cell disease: a report from the Cooperative Study of Sickle Cell Disease. Am J Neuroradiol. 1996;17:965-972.
8. Kinney TR, Sleeper LA, Wang W, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics. 1999;103:640-645.
9. White DA, Moinuddin A, McKinstry RC, et al. Cognitive screening for silent cerebral infarction in children with sickle cell disease. J Pediatr Hematol Oncol. 2006;28:166-169.
10. Hord J, Byrd R, Stowe L, et al. Streptococcus pneumonie sepsis and meningitis during the penicillin prophylaxis era in children with sickle cell disease. J Pediatr Hematol Oncol. 2002;24:470-472.
11. Buchanan GR, DeBaun MR, Quinn CT, Steinberg MH. Sickle cell disease. Hematology Am Soc Hematol Educ Program. 2004;35-37.
12. Powars D, Wilson B, Imbus C, et al. The natural history of stroke in sickle cell disease. Am J Med.
13. Pegelow CH, Adams RJ, McKie V, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126:896-899.
14. Kratovil T, Bulas D, Driscoll MC, et al. Hydroxyurea therapy lowers TCD velocities in children with sickle cell disease. Pediatr Blood Cancer. 2006;47:894-900.
15. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in doppler ultrasonography. N Engl J Med. 1998;339:5-11.
16. Wong WY, Powars DR. Overt and incomplete (silent) cerebral infarction in sickle cell anemia: diagnosis and management. Hematol Oncol Clin North Am. 2005;99:839-855.
17. Adams RJ. Stroke prevention and treatment in sickle cell disease. Arch Neurol. 2001;58:565-568.
18. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317-1322.
19. Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood. 2005;106:2269-2275.
20. Zimmerman SA, Schultz WH, Davis J, et al. Sustained long-term hematologic efficacy of hydroxy urea at maximum tolerated dose in children with sickle cell disease. Blood. 2004;103:2039-2045.
21. Anderson N. Hydroxyurea therapy: improving the lives of patients with sickle cell disease. Pediatr Nurs. 2006;32:541-543.
22. Fathallah H, Atweh GF. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006:58-62.
23. Rodriguez GI, Kuhn JG, Weiss GR, et al. A bioavailability and pharmacokinetic study of oral and intravenous hydroxyurea. Blood. 1998;91:1533-1541.
24. Hydrea (hydroxyurea capsules, USP) package insert. Princeton, NJ: Bristol-Meyers Squibb Company; December 2006.
25. Hankins JS, Helton KJ, McCarville MB, et al. Preservation of spleen and brain function in children with sickle cell anemia treated with hydroxyurea. Pediatr Blood Cancer. 2008;50:293-297.
26. Zimmerman SA, Schultz WH, Burgett S, et al. Hydroxyurea therapy lowers transcranial doppler flow velocities in children with sickle cell anemia. Blood. 2007;110:1043-1047.
27. Wang W, Helms RW, Lynn HS, et al. Effect of hydroxyurea on growth in children with sickle cell anemia: results of the HUG-KIDS study. J Pediatr. 2002;140:225-229.
28. Kinney TR, Helms RW, O’Branski EE, et al. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Blood. 1999;94:1550-1554.
29. Wang WC, Wynn LW, Rogers ZR, et al. A two-year pilot trial of hydroxyurea in very young children with sickle-cell anemia. J Pediatr. 2001;139:790-796.
30. Report of the National Heart, Lung, and Blood Advisory Council Subcommittee Review of the NHLBI Sickle Cell Disease Program. February 29, 2008.
www.nhlbi.nih.gov/resources/
docs/scd_program.htm. Accessed October 1, 2008.
31. Adams R. Big strokes in small persons. Arch Neurol. 2007;64:1567-1574.
32. Hydroxyurea to prevent organ damage in children with sickle cell anemia.
http://clinicaltrials.gov/ct2/
33. Development of a screening strategy for community-based adverse drug related events in the emergency department.
http://clinicaltrials.gov/ct2/
34. Stroke with transfusions changing to hydroxyurea (SWiTCH).
http://clinicaltrials.gov/ct2/show/NCT00122980. Accessed December 3, 2008.
To comment on this article, contact rdavidson@jobson.com.



